CPIC 指南註解:desipramine 與 CYP2D6 基因
摘要
三環抗憂鬱劑(TCA) 具有相似的藥物動力學特性,因此將 CPIC 劑量指引應用於 amitriptyline/nortriptyline 和 CYP2C19、CYP2D6 到其他三環類藥物,包括 desipramine,可能是合理的。CPIC 劑量指引更新建議對於 CYP2D6 中間代謝型(Intermediate metabolizer),nortriptyline 劑量減少 25%。對於 CYP2D6 超快速或 弱代謝型,應考慮使用替代藥物。如果需要使用 nortriptyline,對於 CYP2D6 弱代謝型(Poor metabolizer),考慮減少 50% 劑量。該指引自原始發佈以來已更新。請參閱指引註釋以獲取更多信息。
為特定註釋指定基因型或表現型
註釋
此註釋基於CPIC® 指南,針對三環抗憂鬱劑(TCA)與CYP2D6和CYP2C19。
2019年10月更新
CYP2D6基因型到表現型翻譯變更:截至2019年8月,CYP2D6基因型到表現型翻譯在指南(如CPIC和DPWG)和臨床基因檢測實驗室之間存在一些不一致。CPIC最近進行了一項修改的德爾菲項目,以獲得國際CYP2D6專家小組對統一系統的共識,用於翻譯CYP2D6基因型到表現型 更多信息。對CPIC先前系統的修改包括將CYP2D6*10 等位基因的活性分數計算值從0.5降至0.25,並將活性分數為1的表現型分配從正常代謝型(Normal metabolizer)更改為中間代謝型(Intermediate metabolizer)(所有先前和新表現型分組的表格)。
因此,CYP2D6 等位基因功能表和CYP2D6基因型到表現型表中已進行以下更改(請參閱下方表格):
- 雙倍型導致活性分數為1的從CYP2D6 正常代謝型(NM)更改為CYP2D6 中間代謝型(IM)分配。
- 此指南中建議的影響: CYP2D6 IM的建議(起始劑量減少25%)應考慮用於CYP2D6 AS為1的情況(建議強度:可選建議)。本指南的作者正在更新此指南以反映此變更並評估自本指南發布以來的新證據。
- 所有包含CYP2D6*10 等位基因的雙倍型的活性分數已相應更新(活性分數更改以反映CYP2D6*10的較低活性值0.25)。請參閱所有先前和新表現型分組的表格。
- 此指南中建議的影響: 在共識項目之前,重複的功能正常 等位基因與CYP2D6*10 等位基因的組合導致活性分數為2.5,轉換為超快速代謝型(Ultrarapid metabolizer)。CYP2D6*10的較低值0.25導致這些等位基因組合的活性分數為2.25,根據共識項目轉換為正常代謝型(Normal metabolizer)。
2016年12月更新
2016年12月在線提前發布。
- 2016年CPIC指南更新,關於使用藥物基因組學測試在三環抗憂鬱劑(TCAs)劑量中的應用,已由臨床藥物基因體學實施聯盟(CPIC)發表在《Clinical Pharmacology and Therapeutics》期刊中。審查了截至2016年7月的文獻,更新了建議和補充信息。
- 2016年劑量指南更新摘錄:
- 「Amitriptyline和nortriptyline被用作本指南的代表性TCAs,因為大多數藥物基因組學研究都集中在這兩種藥物上。然而,這些研究的結果可能適用於其他TCAs,因為這些藥物具有可比的藥代動力學特性。」
- 「有大量證據將CYP2D6基因型與三環類藥物的副作用和藥代動力學特徵的表型變異性聯繫起來。對具有影響藥物療效和安全性的CYP2D6基因變異的患者進行藥物治療調整,可能會改善臨床結果並降低初始治療失敗率。」
- 「專注於CYP2D6基因型與TCAs藥代動力學參數或治療結果的關聯的研究很少。CYP2D6活性在幼兒期已完全成熟,但兒童的CYP2C19活性可能相對於成人增加。儘管需要進一步的基因組發育研究,但目前缺乏證據表明本指南不能推廣至兒科患者。」
- 該指南包括基於以下內容的TCAs劑量建議:
- CYP2D6 表現型(下表1)
- 下載並閱讀:
表1:基於CYP2D6 表現型的TCAs劑量建議:
改編自2016年指南更新的表1和表2。
| 可能的表現型 | 活性分數 | 基因型 | 雙倍型的例子 | 影響 | 治療建議a, b | 其他TCAs的建議分類c |
|---|---|---|---|---|---|---|
| CYP2D6 超快速代謝型(Ultrarapid metabolizer)(約1-20%的患者)d | >2.0 | 攜帶多於兩個功能性等位基因的個體 | *1/*1xN, *1/*2xN | 與正常代謝型(Normal metabolizer)相比,TCAs代謝為活性較低的化合物的代謝增加。活性藥物的血漿濃度降低將增加藥物治療失敗的可能性。 | 避免使用三環類藥物,因為可能缺乏療效。考慮使用不經CYP2D6代謝的替代藥物。 如果需要使用TCA,考慮將目標劑量提高(相較於正常代謝型(Normal metabolizer))。利用治療藥物監測(TDM)指導劑量調整。e |
可選建議 |
| CYP2D6 正常代謝型(Normal metabolizer)(約72-88%的患者)d | 1.0-2.0f | 攜帶兩個功能正常 等位基因或兩個功能減弱 等位基因或一個正常和功能缺失 等位基因或一個正常和功能減弱 等位基因或重複等位基因組合,導致活性分數為1.0-2.0的個體 | *1/*1, *1/*2, *2/*2, *1/*9, *1/*41, *41/*41, *1/*4, *1/*5 | TCAs的正常代謝。 | 以推薦的起始劑量開始治療。g | 強烈建議 |
| CYP2D6 中間代謝型(Intermediate metabolizer)(約1-13%的患者)d | 0.5 | 攜帶一個降低和一個功能缺失 等位基因的個體 | *4/*41, *5/*9, *4/*10 | 與正常代謝型(Normal metabolizer)相比,TCAs代謝為活性較低的化合物的代謝減少。活性藥物的血漿濃度較高將增加副作用的可能性。 | 考慮將推薦的起始劑量減少25%。g 利用治療藥物監測(TDM)指導劑量調整。e | 可選建議 |
| CYP2D6 弱代謝型(Poor metabolizer)(約1-10%的患者)d | 0 | 僅攜帶功能缺失 等位基因的個體 | *4/*4, *4/*4xN, *3/*4, *5/*5, *5/*6 | 與正常代謝型(Normal metabolizer)相比,TCAs代謝為活性較低的化合物的代謝大幅減少。活性藥物的血漿濃度較高將增加副作用的可能性。 | 避免使用三環類藥物,因為可能會有副作用。考慮使用不經CYP2D6代謝的替代藥物。 如果需要使用TCA,考慮將推薦的起始劑量減少50%。g 利用治療藥物監測(TDM)指導劑量調整。e |
可選建議 |
a 對於三級胺(例如,amitriptyline),如果CYP2C19基因型結果也可用,請參閱表2中的CYP2C19基於劑量建議和下表3中的CYP2D6/CYP2C19基於劑量建議。
b 劑量建議僅適用於治療抑鬱症等疾病的較高初始劑量的TCAs。請參閱指南中的其他考量,了解用於較低初始劑量的疾病(如神經性疼痛)的劑量建議。
c 建議分類的評分方案在補充中描述。將阿米替林的建議應用於其他也由CYP2D6代謝的TCAs(包括clomipramine、desipramine、doxepin、imipramine 和 trimipramine)可能是合理的。與amitriptyline或nortriptyline相比,這些藥物的基因型指導劑量調整的臨床和藥代動力學數據較少(補充表S8-S16)。
d CYP2D6代謝者狀態頻率基於平均多民族頻率。請參閱CYP2D6頻率表,了解特定人群的等位基因和表現型頻率。
e 根據症狀改善和最小(如果有的話)副作用觀察到的臨床反應來調整劑量。
f 活性分數為1.0的患者可能被某些參考實驗室分類為中間代謝型(Intermediate metabolizer)。
g 患者可能會接受三環類藥物的初始低劑量,然後在幾天內增加到推薦的穩態劑量。本指南中的起始劑量指的是推薦的穩態劑量。
2013年5月
關於三環抗憂鬱劑(TCA)劑量中使用藥物基因組學測試的指南已由藥物基因體學臨床應用聯盟(CPIC)發表在《Clinical Pharmacology and Therapeutics》期刊中。
劑量指南摘錄:
Amitriptyline 和 nortriptyline 被用作本指南的模型藥物,因為大多數藥物基因組學研究都集中在這兩種藥物上。由於三環類藥物具有可比的藥代動力學特性,將此指南應用於其他三環類藥物(包括clomipramine(補充表S15))可能是合理的,但需承認這些藥物的劑量調整支持數據少於 amitriptyline 或 nortriptyline。
請參閱 nortriptyline,了解總結 CYP2D6 和 CYP2C19 基於的劑量建議的摘錄和表格,當需要較高的初始起始劑量時(文章)。
Sympathetic Nerve 途徑 (神經效應接合處)
概括
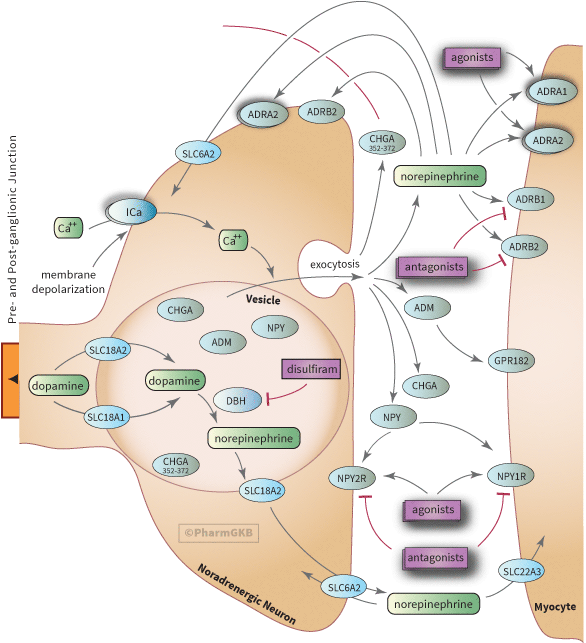
簡化的交感神經效應接頭示意圖,顯示可能參與的基因。
描述
該圖示描繪了一個傳出型 Sympathetic Nerve(後神經節交感神經元)。這類神經元由來自胸腰脊髓的前神經節纖維所支配。前神經節與後神經節交感神經纖維的突觸連接發生在椎旁神經節(或稱「交感神經鏈」)。
前神經節軸突釋放神經傳導物質乙醯膽鹼(ACh)。乙醯膽鹼的合成由膽鹼乙酰轉移酶(ChAT)催化,該酶在突觸前神經末梢中從乙酰輔酶A和膽鹼合成。合成過程的速率受到膽鹼通過神經末梢的轉運蛋白的限制 (SLC5A7)。第二個轉運蛋白 (SLC18A3) 負責將乙醯膽鹼攝取到神經末梢的突觸囊泡中。囊泡釋放依賴鈣,並被認為是由囊泡與神經末梢膜在外排過程中融合所引起的。ACh 擴散穿過突觸間隙到達後神經節神經元的細胞體,並激活尼古丁型膽鹼受體(nAChRs),這些受體是由外部配體 ACh 控制的五聚體陽離子通道。在交感神經元中,這些 nAChRs 可以是異源五聚體或同源五聚體。交感神經元的異源五聚體通常由兩個 alpha-3 亞基 (CHRNA3) 和三個 beta-4 亞基 (CHRNB4) 組成。這類神經元也具有同源五聚體的 CHRNA7 受體。
在中樞神經系統中,尼古丁型受體通常是 alpha-4 (CHRNA4) 和 beta-2 (CHRNA2) 亞基的異源五聚體組合,或是 alpha-7 同源五聚體 (CHRNA7)。
乙醯膽鹼酯酶 (ACHE) 由單一基因編碼,具有多種剪接選項,負責在突觸中將乙醯膽鹼降解為膽鹼和醋酸(主要在突觸間隙中,雖然在突觸前末梢也有一些)。膽鹼和醋酸均無顯著的效能,因此水解過程終止了傳遞物質的作用。
尼古丁型受體的激活會打開中央陽離子孔,ADM 使 Na+ 或 Ca++(或兩者)進入細胞內部。這種陽離子的進入可以使細胞膜去極化,從而打開電壓依賴的 Ca++ 通道,觸發外排作用並激活轉錄。
兒茶酚胺的生物合成始於酪氨酸(或苯丙氨酸),並經過 norepinephrine(去甲腎上腺素)。兒茶酚胺生物合成中的限速酶是酪氨酸羥化酶(TH),其受到轉錄和翻譯後控制。最終的兒茶酚胺生物合成步驟 (DBH) 發生在兒茶酚胺儲存囊泡內;其他生物合成酶則位於細胞質中。關鍵的酶輔因子包括:TH:四氫生物喹啉,Fe++。DBH:抗壞血酸,Cu++。
兒茶酚胺通過 VMATs 被運輸到儲存囊泡中,特別是神經元中的 SLC18A2 (VMAT2),但在嗜鉻細胞中也有 SLC18A1 (VMAT1)。
在 norepinephrine 釋放後,傳遞物質會通過兩個過程被移除或攝取:
「攝取-1」:通常是神經元的高親和力、立體選擇性、依賴 Na+ 的過程,由 SLC6A2 媒介。
「攝取-2」:通常是非神經元的,親和力較低。由有機陽離子轉運蛋白家族,特別是 SLC22A3 (OCT3) 媒介。
兒茶酚胺也會在神經和非神經部位進行代謝。在神經中,途徑 包括單胺氧化酶 (MAOA 和 MAOB) 以及兒茶酚-O-甲基轉移酶 (COMT)。
幾種神經肽與兒茶酚胺共同儲存並共同釋放,來自 Sympathetic Nerve 的大型致密核心兒茶酚胺儲存囊泡,包括色素顆粒蛋白 A (CHGA), 神經肽 Y (NPY) 和腎上腺髓質素 (ADM,特別是來自腎上腺細胞)。 CHGA 是內源性尼古丁型膽鹼拮抗肽「catestatin」 (CHGA 352-372) 的來源。
在腎上腺髓質的嗜鉻細胞以及 epinephrinergic 神經元中,另一個兒茶酚胺生物合成步驟發生:PNMT(苯乙醇胺 N-甲基轉移酶),其將 norepinephrine(去甲腎上腺素) N-甲基化形成 epinephrine(腎上腺素)。 PNMT 發生在細胞質中。
在 dopaminergic 軸突(神經)中,兒茶酚胺生物合成在 dopamine 結束,該物質被釋放並受到 dopamine 轉運蛋白的再攝取 (DAT, SLC6A3)。
在血清素能軸突(神經)中,傳遞物質生物合成的限速酶是色氨酸羥化酶,傳遞物質的合成以血清素(5HT,5-羥基色胺)結束,該物質被釋放並受到血清素轉運蛋白的再攝取 (SERT, SLC6A4)。
Imipramine/Desipramine 途徑, 藥物動力學
概括
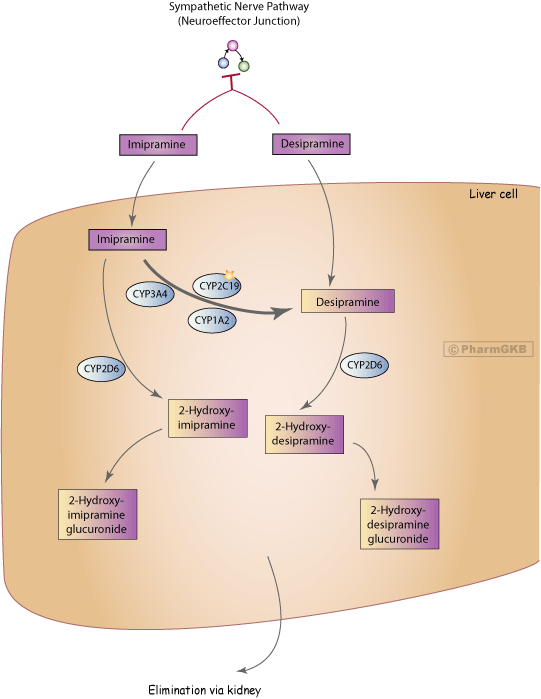
參與 imipramine 和 desipramine 代謝之候選基因示意圖
描述
隨著選擇性血清素再攝取抑制劑 (選擇性血清素再回收抑制劑,SSRIs) 的引入,三環抗憂鬱劑 (TCAs) 在治療抑鬱症方面的主要使用已減少[Article:10319193]。SSRIs 具有更可耐受的副作用特徵及較低的毒性風險[Article:16553509]。然而,TCAs 仍然是某些類型抑鬱症的有效治療選擇,例如伴有憂鬱特徵的重度抑鬱症及中風後抑鬱症[Articles:8835647,12893112]。TCAs 也被認為對纖維肌痛、神經性疼痛及偏頭痛預防有益[Articles:11029681,12230591,15336464]。desipramine 在治療成人注意力不足過動症方面似乎有效[Article:16732717]。
經口給藥後,TCAs 迅速被吸收。TCAs 是脂溶性化合物,因此顯示出較大的分佈容積。它們與血漿白蛋白及外血管組織結合[Article:10319193]。TCAs 在 2-6 小時內達到血漿濃度峰值。它們經過 P450 同工酶的廣泛肝臟代謝,且少於 5% 的 TCA 劑量以未改變的形式排除[Article:10319193]。
CYP2C19 活性與三級胺 TCA 的 N-去甲基化有關。P450 同工酶 CYP1A2 和 CYP3A4 也參與三環類藥物的去甲基化[Articles:10319902,9084901,17471183]。三級胺 imipramine 通常通過去甲基化代謝為次級活性代謝物 desipramine。desipramine 本身也是一種三環藥物。三級和次級胺主要通過 CYP2D6 的羥基化代謝為 2-OH- imipramine 和 2-OH-desipramine [Articles:10319193,16553509]。TCAs 的羥基代謝物具有與其母體化合物相似或更短的半衰期及較小的分佈容積[Articles:7461025,2687861]。TCA 羥基化代謝物的葡萄糖醛酸結合改善了其水溶性,因此促進了更有效的腎臟排泄。然而,羥基代謝物也以未結合形式排入尿液[Article:2116227]。通常,TCAs 的清除率被觀察到與藥物濃度成正比,但一些研究顯示在高劑量的 imipramine 和 desipramine 下,P450 的羥基化出現飽和的劑量依賴性動力學[Articles:1813901,10319193,3709631,6467794,2687860]。
許多 三環抗憂鬱劑(TCA) 的劑量範圍為 25 - 300 mg/天。基因差異在 CYP2C19 和 CYP2D6 的活性中在很大程度上解釋了藥物代謝和血漿濃度的個體間變異[Article:10319193]。根據抗抑鬱藥的不同,CYP2C19 弱代謝型(Poor metabolizer),以及 CYP2D6 弱代謝型(Poor metabolizer),在常規劑量下可能產生幾倍於正常的血漿藥物水平[Articles:15037866,16384813]。在 TCA 的血漿濃度超過 450-500 ng/ml 時,會出現如焦躁、混亂和徘徊等毒性效應[Article:9139291]。主要毒性和死亡與濃度超過 1000 ng/ml 有關[Articles:17471183,9139291]。在藥物相互作用方面,desipramine 不是 強烈建議 的 CYP2D6 抑制劑,但 imipramine 顯著抑制 CYP2C19 和 CYP1A2 [Article:17471183]。