CPIC 指南註解:ivacaftor 與 CFTR 基因
摘要
建議僅在囊性纖維化(Cystic fibrosis, CF)(CF)患者中使用Ivacaftor治療,這些患者必須是同型合子(Homozygous)或異型合子(Heterozygous)某些CFTR變異的攜帶者。請參閱完整指南以獲取免責聲明、更多詳細信息和支持證據。該指南自原始發佈以來已更新。請參閱指南註釋以獲取更多信息。
為特定註釋指定基因型或表現型
註釋
此註釋基於CPIC® 指南,針對 ivacaftor 和 CFTR。
2025年8月更新
- FDA核准的藥物仿單 ivacaftor 已更新,新增58個變異,總計96個指示變異:A120T, A234D, A349V, D192G, D924N, E822K, F311del, F311L, F508C, G1249R, G178E, G194R, G314E, G576A, G970D, H1375P, H939R, I1027T, I1139V, I148T, I175V, I807M, L1480P, L320V, L967S, L997F, M152V, M952I, M952T, Q1291R, Q237E, Q237H, Q359R, R1162L, R117G, R117L, R117P, R1283M, R170H, R347L, R553Q, R668C, R75Q, R792G, R933G, S1159F, S1159P, S589N, T1053I, T338I, V1293G, V232D, V562I, V754M, W1282R, Y1014C, Y1032C。下表1已更新。然而,上述特定變異未在2014年指南出版中討論。
2019年5月更新
- 自2014年4月CPIC ivacaftor劑量指南的最新更新,以及2017年6月的最新線上更新以來,FDA核准的藥物仿單再次更新,新增5個CFTR變異,核准用於KALYDECO,總計38個指示變異:2789+5G->A (rs80224560), 3272-26A->G (rs76151804), 3849+10kbC->T (rs75039782), 711+1G->T (rs77188391) 和 E831X (rs78655421)。下表1已更新。
2017年6月更新
- 自2014年4月CPIC ivacaftor劑量指南的最新更新,以及2016年5月的最新線上更新以來,FDA核准的藥物仿單再次更新,新增23個CFTR變異,核准用於KALYDECO,總計33個指示變異。請參閱2017年5月17日的FDA新聞稿以獲取更多信息。鑑於此變更,CPIC指南註釋在此網頁上已更新以包含這些額外的CFTR變異(見下表1)。這些變異未在2014年指南出版中討論。這些變異如下:E56K (rs397508256), P67L (rs368505753), R74W (rs115545701), D110E (rs397508537), D110H (rs113993958), R117C (rs77834169), E193K (rs397508759), L206W (rs121908752), R347H (rs77932196), R352Q (rs121908753), A455E (rs74551128), D579G (rs397508288), S945L (rs397508442), S977F (rs141033578), F1052V (rs150212784), K1060T (rs397508513), A1067T (rs121909020), G1069R (rs200321110), R1070Q (rs78769542), R1070W (rs202179988), F1074L (rs186045772), D1152H (rs75541969), D1270N (rs11971167).
2016年5月更新
- 自2014年4月CPIC ivacaftor劑量指南的最新更新以來,FDA核准的藥物仿單再次更新,新增變異R117H (rs78655421)。鑑於此變更,CPIC指南註釋在此網頁上已更新以包含此額外的CFTR變異(見下表1)。此變異未在2014年指南出版中討論。此外,更新的藥物仿單指示ivacaftor用於2歲及以上患者;之前僅指示用於6歲及以上患者。
2014年4月更新
- 在提交和審查CPIC指南手稿後,FDA核准的藥物仿單 ivacaftor已更新以包含額外的變異。鑑於這些變更,CPIC指南註釋在此網頁上已更新以包含額外的CFTR變異,具體為G1244E (rs267606723), G1349D (rs193922525), G178R (rs80282562), G551S (rs121909013), S1251N (rs74503330), S1255P (rs121909041), S549N (rs121908755) 和 S549R (rs121908757 和 rs121909005)(見下表1)。這些變異未在2014年指南出版中討論。
2014年3月
接受文章預覽線上2014年3月;提前線上出版2014年3月。
- 關於使用藥物基因組學測試來決定是否應進行ivacaftor治療的指南已由臨床藥物基因體學s實施聯盟(CPIC)發表於《Clinical Pharmacology and Therapeutics》期刊。
- 這些指南適用於
- 囊性纖維化(Cystic fibrosis, CF)患者
- 兒科,6歲及以上
- 成人
- 下載並閱讀:
表1:基於CFTR基因型的ivacaftor推薦治療使用
_改編自2014年指南手稿和線上更新的表2。下表中新增了未在已發表的2014年指南或補充中出現的變異;特別是G551D和F508del以外的變異。
| CFTR基因型 | 雙倍型的例子 | 對ivacaftor效果的影響 | ivacaftor治療的建議 | ivacaftor治療的建議分類c |
|---|---|---|---|---|
| 同型合子(Homozygous)或異型合子(Heterozygous) G551D-CFTR, rs75527207基因型AA或AG | G551D/ F508del, G551D/ G551D | 顯著改善肺功能、體重、肺部惡化風險、患者報告的結果,以及通過增強CFTR通道活性(增加開放通道的概率)降低汗氯濃度。 | 根據產品仿單使用ivacaftor | 強烈建議 |
| 同型合子(Homozygous)對於F508del-CFTR, rs113993960或rs199826652基因型del/del | F508del/F508del | 汗氯濃度無顯著降低;其他臨床測量無變化,包括肺功能測量、肺部惡化或體重b。不太可能對治療有反應。 | 不建議使用ivacaftora | 中等建議b |
| 同型合子(Homozygous)或異型合子(Heterozygous)對於以下CFTR變異之一:E56K, P67L, R74W, D110E, D110H, R117C, R117H, G178R, E193K, L206W, R347H, R352Q, A455E, S549N, S549R, G551D, G551S, D579G, S945L, S997F, F1052V, K1060T, A1067T, G1069R, R1070Q, R1070W, F1074L, D1152H, G1244E, S1251N, S1255P, D1270N, G1349D, 2789+5G->A, 3272-26A->G, 3849+10kbC->T, 711+1G->T 和 E831X, A120T, A234D, A349V, D192G, D924N, E822K, F311del, F311L, F508C, G1249R, G178E, G194R, G314E, G576A, G970D, H1375P, H939R, I1027T, I1139V, I148T, I175V, I807M, L1480P, L320V, L967S, L997F, M152V, M952I, M952T, Q1291R, Q237E, Q237H, Q359R, R1162L, R117G, R117L, R117P, R1283M, R170H, R347L, R553Q, R668C, R75Q, R792G, R933G, S1159F, S1159P, S589N, T1053I, T338I, V1293G, V232D, V562I, V754M, W1282R, Y1014C, Y1032Cd | F508del/S549N | 顯著增強通道開放概率體外 [文章:22293084]。體外使用CFBEo-細胞表達S549N-CFTR的測試顯示ivacaftor增強氯通道功能[文章:23027855],且一個病例研究顯示在一名12歲患有CF且帶有S549N變異的女孩中,ivacaftor治療後肺功能改善[文章:24081349]。在帶有R117H變異的患者中,汗氯和CFQ-R呼吸域得分改善[文章:26070913]。或者,變異被列在FDA核准的藥物仿單中,對ivacaftor有反應。 | 根據產品仿單使用ivacaftor | 中等建議 |
a這些建議基於單獨使用ivacaftor治療CF患者和當前證據。目前正在進行臨床試驗以研究單獨使用ivacaftor或與其他藥物聯合使用以治療帶有G551D以外CFTR變異的CF患者,因此ivacaftor可能對這些患者有效。詳情請參閱2014年指南。
b對於F508del/F508del基因型患者的建議基於ivacaftor的作用機制和臨床觀察數據。然而,臨床研究是一項安全性研究,並未設計以檢測療效差異[文章:22383668]。
c評分方案描述於2014年補充中。
d此表中列出的變異包括新增至更新的藥物仿單 ivacaftor中的變異。此表的修改是在2014年CPIC Ivacaftor-CFTR指南接受出版後進行的[文章:24598717],並未反映在CPIC指南主手稿或補充的PDF中。
e不建議對其他CFTR變異或同型合子(Homozygous)對F508del變異的CF患者使用ivacaftor(詳情、支持證據和免責聲明請參閱2014年指南)。其他CFTR變異的未來臨床試驗正在進行中。
Ivacaftor and Lumacaftor 途徑, 藥物動力學/藥效學
概括
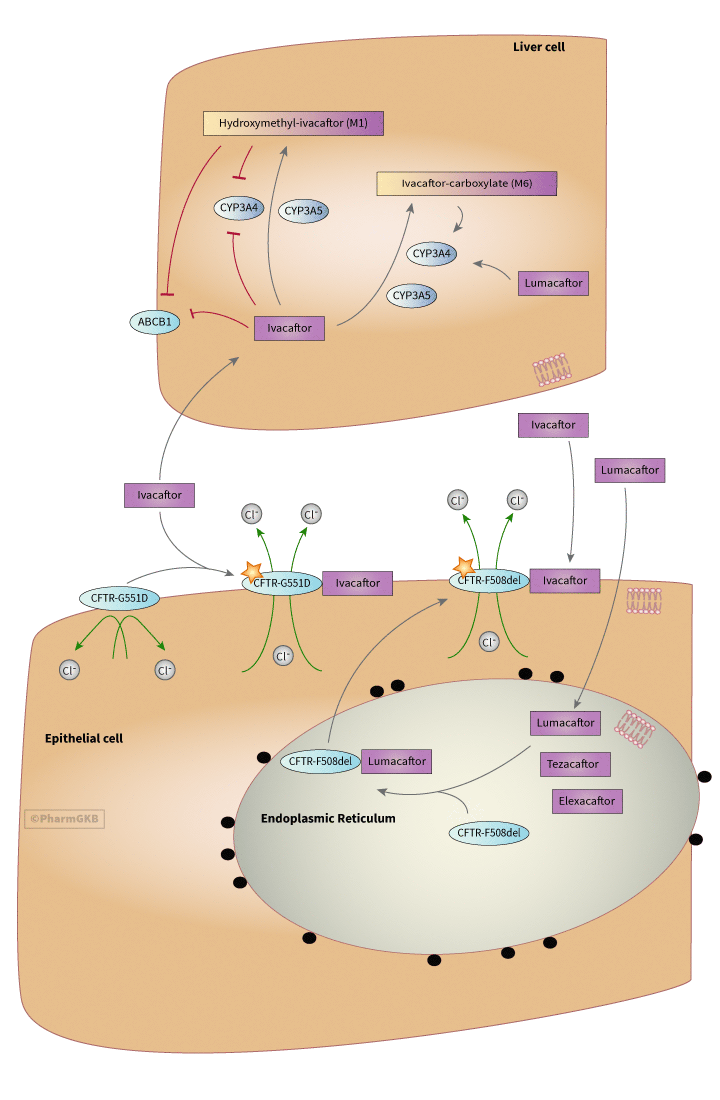
示意圖展示了肝細胞中參與 Ivacaftor 代謝的相關基因,以及上皮細胞在單獨使用 Ivacaftor,或合併使用 Ivacaftor 與 Lumacaftor 治療下,CFTR 蛋白門控功能增強的情形。
描述
引言
ivacaftor 是最早開發的藥物之一,旨在治療 囊性纖維化(Cystic fibrosis, CF) (CF) 的根本原因,而非僅僅緩解症狀。它是一種小分子增效劑,能夠增強 擇期 跨膜電導調節蛋白 (CFTR) 的功能。增效劑是一類新型的 CFTR 調節劑,旨在恢復 CFTR 功能 [Article:24656117]。美國食品藥品監督管理局 (FDA) 於 2012 年批准了 ivacaftor,因其適用於特定 CFTR 變異的患者,特別是類別 III 變異。美國約有 30,000 人患有 CF,這一數字低於美國孤兒藥物的 200,000 患者門檻 [Article:24656117]。單獨使用 ivacaftor 並未獲得對 F508del 的批准,該變異是 CFTR 中最常見的變異 USA [Article:23616732]。額外的調節劑 lumacaftor、elexacaftor 和 tezacaftor 被設計為對 F508del 有效。
CF 是一種常染色體隱性遺傳疾病,其特徵是汗液 chloride 濃度超過 60 mmol/L,導致進行性阻塞性肺病和早期死亡,以及肝臟、胰腺、輸精管和腸道的問題 [Article:24534272]。CF 在全球約影響 70,000 人,包括美國每 3,500 名新生兒中約有 1 名 [Article:23457166,其病因是繼承了兩個有害的 CFTR 基因 (囊性纖維化(Cystic fibrosis, CF) 基金會)。CFTR 蛋白質是一種 chloride 通道,維持細胞內外的離子和水的平衡 [Article:24561283]。該通道通過 ATP 結合、水解和磷酸化來開啟和關閉,這改變了蛋白質的構象,使 chloride 離子能夠在細胞內外區域之間的離子梯度中流動 [Article:24534272]。缺陷的 CFTR 功能阻止了 chloride 離子穿過上皮細胞的流動,導致粘液積聚、感染、炎症以及逐漸減少的肺功能,這是 囊性纖維化(Cystic fibrosis, CF) 的特徵 [Article:24561283]。
藥物動力學
作為口服劑量給藥,ivacaftor 在腸道中易於吸收,但在水中的溶解度較低(< 0.05 ug/mL)人用藥品委員會,Kalydeco:評估報告。2012,歐洲藥品管理局。服用 150 mg 劑量的 ivacaftor 與高脂餐一起可改善吸收,增加 AUC 2.5 倍,並將 Tmax 從 3 小時延遲至 5 小時 [Article:26968005。劑量/時間 藥物動力學 在 250 mg 劑量下遵循線性曲線,但 Cmax 在 375 mg 及以上劑量時達到平穩 人用藥品委員會,Kalydeco:評估報告。2012,歐洲藥品管理局]。ivacaftor 在血漿中以高度結合(99%)的形式運輸,優先結合於 α-1-酸性糖蛋白,但也結合於白蛋白,作用位點為上皮細胞的頂膜 [Articles:26968005,25148205。然而,預期不會有與蛋白質結合競爭相關的 藥物交互作用 人用藥品委員會,Kalydeco:評估報告。2012,歐洲藥品管理局]。 如圖所示,ivacaftor 在肝臟中由細胞色素 P450 3A (CYP3A) 代謝,包括 CYP3A4 和 CYP3A5,生成一種代謝物羥基甲基-ivacaftor (M1),該代謝物被認為是活性的,其效力為 ivacaftor 本身的 1/6,還有一種非活性代謝物 ivacaftor-羧酸鹽 (M6),該代謝物不被認為是活性的,其活性水平為 ivacaftor 的 1/50 [Article:25103957]。母藥和代謝物的排泄主要通過膽汁進行(87%),其中 22% 為 M1 代謝物,43% 為 M6 代謝物 [Article:25148205]。ivacaftor 的半衰期為 12-14 小時 人用藥品委員會,Kalydeco:評估報告 2012,歐洲藥品管理局。
ivacaftor 和 M1 被認為是 CYP3A4 的弱抑制劑,增加了中氟噻噴在血漿中隨時間變化的藥物濃度曲線面積 (AUC) 54% [Article:25103957。它們也是 P-糖蛋白 (ABCB1] ) 的弱抑制劑,增加了地高辛 AUC 32% [Article:25103957]。CYP3A 誘導劑,如利福平、卡馬西平和苯妥英,會降低對 ivacaftor 的暴露,而 CYP3A 抑制劑則會增加對 ivacaftor 的暴露 [Articles:25103957,26581802]。當與已知的 CYP3A 抑制劑一起給藥時,建議將 ivacaftor 的給藥頻率從每日兩次減少為每日一次 人用藥品委員會,Kalydeco:評估報告 2012,歐洲藥品管理局。
藥效學
ivacaftor 適用於攜帶至少一種 11 種經批准的基因變異的 CF 患者,這些變異影響 CFTR 的閘控能力,其中大多數為類別 III 變異,這些變異影響 chloride 通道的激活,從而抑制正常的 chloride 運動 [Articles:21083385, 12503104]。這些變異列在表中。類別 III 變異的功能後果意味著 CFTR 正常定位於頂膜,但無法進行 cAMP 介導的激活,因此是非功能性的 [Articles:12503104, 26390337]。ivacaftor 是一種 擇期 增效劑,據信能穩定通道的開啟狀態,使 chloride 運輸得以進行 [Article:23440202]。這種增效作用的具體機制尚不清楚,但可能是通過解耦閘控循環和 ATP 水解循環,或通過增加 ATP- 依賴的開啟速率和減慢關閉速率來實現 [Articles:23440202, 24796242]。據信通過結合 CFTR 在上皮細胞膜中,ivacaftor 改善了 CFTR 的功能,該蛋白質具有閘控突變,並且 CFTR 具有 功能正常 [Articles:25148205, 23440202]。 最初的驗證研究是在 CF 患者中進行的,這些患者至少有一種 G551D CFTR 閘控變異(c.1652G>A, rs75527207);治療報告顯示肺功能改善超過 10%,足以顯著改善 CF 症狀 [Article:22293084]。ivacaftor 治療報告顯示,在 24 週內,強制呼氣量(FEV-1)的肺功能測試改善了 10.4% - 17.5%,並且這一差異在 48 週的試驗結束時得以維持 [Articles:24461666, 22047557]。ivacaftor 治療報告顯示,體重增加(在 ivacaftor 上增加 3.7 kg,相較於安慰劑的 1.8 kg,為期 24 週)並且與基線相比,chloride 濃度下降(-55.5 mmol/L ivacaftor 相較於安慰劑的 -1.8 mmol/L)在 6-11 歲的臨床試驗中 [Article:24461666]。汗液 chloride 濃度的變化通常用作 囊性纖維化(Cystic fibrosis, CF) 藥物生物活性的生物標誌物,儘管對此測量的有效性存在爭議 [Articles:23276841, 24258833, 24660233]。在 12 歲及以上的患者中報告了 chloride 運輸的改善 [Article:22047557]。在與安慰劑相比,ivacaftor 治療 2 週後報告了呼吸改善 [Article:22047557]。
對於 F508del 變異(c.1521_1523delCTT; rs113993960 或 rs199826652)的患者進行的試驗顯示,該變異並非閘控變異,而是阻止 CFTR 蛋白質從內質網排出並定位到細胞膜,單獨使用 ivacaftor 治療未顯示改善 [Article:19846789]。然而,ivacaftor 與 lumacaftor 的聯合藥物 (VX-809) 於 2015 年獲得 FDA 和歐洲藥品管理局 (EMA) 的批准,用於 同型合子(Homozygous) 的 F508del 等位基因 患者 [Article:26417173]。lumacaftor 報導恢復了來自 同型合子(Homozygous) 的 F508del 等位基因 的下呼吸道上皮細胞中約 15% 的正常 CFTR 功能,通過輔助蛋白折疊 [Article:26581802]。通過改善由該 等位基因 引起的折疊故障,lumacaftor 被認為能夠使 F508del CFTR 定位到細胞膜 [Article:26581802]。當 lumacaftor 與 ivacaftor 結合時,來自 同型合子(Homozygous) 的 F508del CFTR 的折疊修復與 ivacaftor 的增效作用相結合,報導顯示在下呼吸道上皮細胞中功能改善 30% [Article:26581802]。第二和第三期試驗也顯示在 同型合子(Homozygous) 的 F508del 等位基因 患者中具有臨床意義的療效,結果通過 chloride 濃度和 FEV-1 的變化來衡量 [Article:26581802]。額外的修飾劑 tezacaftor 和 elexacaftor 針對 F508del 變異 [Article:38249954]。大約 90% 的 囊性纖維化(Cystic fibrosis, CF) 患者符合 ivacaftor 含有的療法 [Article:38249954]。
藥物基因體學
CFTR 具有兩個核苷酸結合域和兩個膜穿透域,變異可以發生在所有區域 [Article:21652558]。從高爾基體運輸到頂膜的 CFTR 在分泌囊泡中以每分鐘 10% 的速度更新,半衰期為 12-24 小時 [Article:21652558]。在 CFTR 基因中已識別出超過 2000 種變異,但只有 127 種已被確認為致病性。美國醫學遺傳學會的指導方針將這 23 種變異納入其推薦的面板中,以確定 囊性纖維化(Cystic fibrosis, CF) 的攜帶者狀態 [Article:21422883]。 在 CFTR 中的致病變異根據其對 CFTR 功能的影響分為 5 類。類別 I 變異影響 CFTR 的生物合成,產生截短或不穩定的蛋白質,迅速被降解。類別 II 變異影響蛋白質折疊,可能降低蛋白質的穩定性和適當定位。F508del 變異是一種類別 II 變異,阻止定位到細胞膜,出現在超過 90% 的 CF 病例中 [Article:24534272]。類別 III 變異,即那些被 ivacaftor 針對的變異,能夠正確折疊和定位,但無法被 ATP 或磷酸化調節,因此不具閘控功能。類別 IV 變異也影響離子流,但通過降低 chloride 離子的通透性,而類別 V 變異通常導致剪接變化,減少 CFTR 在細胞膜中的表達 [Article:21652558]。 在 ivacaftor 治療中最重要的基因變異是類別 III 變異,這些變異在 CFTR 中造成閘控問題。G551D 變異是最常見的閘控變異,據認為在約 4-5% 的 CF 患者中存在 [Article:21083385]。FDA 和 EMA 最初於 2012 年在美國和歐洲批准使用 ivacaftor 來治療由 G551D 閘控變異引起的 囊性纖維化(Cystic fibrosis, CF) [Article:25148205]。自那以後,批准範圍已擴大至包括其他閘控變異。經批准使用 ivacaftor 的變異列表見表中,包括 G1244E、G1349D、G178R、G551S、S1251N、S1255P、S549N 和 S549R [Articles:25148205, 22293084]。這些變異在 囊性纖維化(Cystic fibrosis, CF) 患者中各自的發生率均低於 1% [Articles:24561283, 22293084]。除了這些其他類別 III 變異外,FDA 還批准了 ivacaftor 用於治療具有 R117H 變異的患者,這是一種主要為導電變異的類別 IV 變異,但也具有缺陷的閘控活性 [Article:26324139]。
經批准的 CFTR 變異,用於治療 ivacaftor
| 氨基酸變化 | rs 編號 | cDNA 變化 |
|---|---|---|
| R117H | rs78655421 | 350G>A/C/T |
| G178R | rs80282562 | 532G>A |
| S549R | rs121908755 | 1646G>A/T |
| S549R | rs121908755; rs121909005 | 1645A>C; 1647T>G |
| G551S | rs121909013 | 1651G>A |
| G551D | rs75527207 | 1652G>A |
| G1244E | rs267606723 | 3731G>A/T |
| S1251N | rs74503330 | 3752G>A |
| S1255P | rs121909041 | 3763T>C |
| G1349D | rs193922525 | 4046G>A |
結論
ivacaftor 單藥治療增強了具有缺陷閘控能力的 CFTR 蛋白質的功能,其中最常見的原因是 G551D 變異。只要在 CFTR 的患者中恢復了定位,ivacaftor 也能改善這些個體的 chloride 運輸。雖然具體的作用機制尚不清楚,但 ivacaftor 被認為是通過 CFTR 恢復 chloride 離子通過細胞膜的流動,從而減少具有閘控缺陷的患者的 CF 症狀。