CPIC 指南註解:dapsone、methylene blue、pegloticase、rasburicase、tafenoquine、toluidine blue 與 G6PD 基因
摘要
高風險藥物應避免用於G6PD 缺乏患者,不論是否患有慢性非球形溶血性貧血(chronic non-spherocytic hemolytic anemia, CNSHA)。在G6PD 可變型(Variable)或無法判定 表現型患者中,應在開始藥物治療前進行酶活性測試。
G6PD基因位於X染色體上。因此,一些患者可能只有一個拷貝,而其他患者可能有兩個拷貝。請參閱完整指南以獲取免責聲明、更多詳情和支持證據。
為特定註釋指定基因型或表現型
註釋
此註釋基於擴展的CPIC®指引,針對G6PD基因型的藥物使用。CPIC作者評估了攜帶G6PD變異的患者使用各種藥物的可用證據。
2022年9月
-
擴展的CPIC®指引,針對G6PD基因型的藥物使用已在《Clinical Pharmacology and Therapeutics》期刊中發表。作為該指引的一部分,作者將藥物按其在G6PD缺乏症患者中引起急性溶血性貧血(AHA)的風險進行分類。Dapsone、methylene blue、pegloticase、rasburicase、tafenoquine和toluidine blue被分類為高風險藥物,如指引出版物的表2所示。此指引取代了2014年原始的G6PD和rasburicase指引。
-
這些指引適用於:
- 成人患者
- 兒科患者
-
指引摘錄:
- 「在此CPIC指引中增加了一個步驟,將藥物分為三組:在G6PD缺乏症的情況下可被視為高風險的藥物(因此通常應避免使用),在G6PD缺乏症中被視為中等風險的藥物(因此應謹慎使用),以及可被視為低至無風險的藥物(在G6PD缺乏症患者中不會增加AHA風險,與正常G6PD狀態者相比)。」
- 「為了將藥物分配到風險組別,作者不僅考慮了主要同行評審文獻中的證據強度,還考慮了藥物使用頻率、監管機構警告的存在以及是否存在可能產生活性氧物質並在G6PD缺乏症中促成溶血的機制(補充材料,分配風險等級)」
- 「Rasburicase和pegloticase(高風險藥物) 在G6PD缺乏症中rasburicase和pegloticase的風險類別基於文獻強度(高水平證據)將AHA和高鐵血紅蛋白血症與在G6PD缺乏症情境下使用這些藥物聯繫起來(表S1),尿酸氧化酶的作用機制(產生過氧化氫)(31),以及強烈建議,一致的監管警告(表S3),rasburicase和pegloticase被認為屬於高風險類別。」
- 「治療建議.作為高風險藥物,rasburicase和pegloticase應避免在G6PD缺乏症患者中使用(表3)。然而,對於任何藥物,必須權衡不良反應(AHA和/或高鐵血紅蛋白血症)的風險與高尿酸血症的風險,特別是在預期腫瘤溶解的剛診斷惡性腫瘤患者中。腫瘤溶解綜合症本身可能危及生命,替代的尿酸降低療法,如allopurinol,可能不如rasburicase有效降低尿酸水平,並且具有其他潛在不良反應。」
-
下載並閱讀:
- 擴展的藥物基因體學臨床應用聯盟(CPIC)指引,針對G6PD基因型的藥物使用
- 2022補充材料
- G6PD 基因資訊表
- Drug Resource Mappings
- Pre and Post Test Alerts
- 高風險藥物流程圖
表1:高風險藥物在G6PD 表現型中的推薦治療使用
改編自指引的表1、2和3
| 預測表現型 | 基因型a | 基因型範例b | 影響 | 治療建議 | 建議等級c | 考量 |
|---|---|---|---|---|---|---|
| 正常 | 攜帶一個X染色體,帶有非缺乏(IV類)等位基因 或攜帶兩個非缺乏(IV類)等位基因s |
B, Sao Boria, IV B/B, B/Sao Boria, B/A, IV/IV |
急性溶血性貧血風險低 | 基於G6PD狀態,無理由避免高風險藥物 | 強烈建議 | Tafenoquine的安全性已在G6PD酶活性≥正常的70%時確立 |
| 缺乏 | 攜帶一個X染色體,帶有缺乏(II-III類)等位基因 或攜帶兩個缺乏(II-III類)等位基因s或一個I類等位基因和一個II或III類等位基因 |
A-, Orissa, Kalyan-Kerala, Mediterranean, Canton, Chatham, II, III A-/A-, A-/Orissa, Orissa/ Kalyan-Kerala, Mediterranean/ Mediterranean, Chatham /Mediterranean, Canton/ Viangchan, II/II, II/III, III/III, I/II, I/III |
急性溶血性貧血風險高 | 避免使用高風險藥物 | Methylene blue和toluidine bluef: 中等建議. 所有其他高風險藥物: 強烈建議 |
|
| 嚴重缺乏合併慢性非球形紅細胞溶血性貧血(Deficient with CNSHA) | 攜帶一個X染色體,帶有缺乏(I類)等位基因 或攜帶兩個缺乏(I類)等位基因sd |
Bangkok, Villeurbanne, I Bangkok/Bangkok, Bangkok/Villeurbanne, I/I |
慢性溶血性貧血急性加重的高風險 | 避免使用高風險藥物 | 強烈建議 | 雖然在G6PD 嚴重缺乏合併慢性非球形紅細胞溶血性貧血(Deficient with CNSHA) 表現型個體中沒有發表的數據,但基於G6PD 缺乏個體的證據,有強烈建議理由避免這些藥物 |
| 可變型(Variable)e | 攜帶一個非缺乏(IV類)等位基因和一個缺乏(I-III類)等位基因 | B /Bangkok, B/Mediterranean, B/A-, IV/I, IV/II, IV/III | 可變型(Variable)急性溶血性貧血風險 | 基於G6PD狀態,無理由在標準劑量下避免低至無風險藥物 | 中等建議 | 由於X 染色體聯鎖嵌合現象(X-linked mosaicism),攜帶多於一個X染色體的個體(例如,女性,Klinefelter綜合症患者)和異型合子(Heterozygous)一個非缺乏(IV類)和一個缺乏(I-III類)等位基因可能顯示正常或缺乏 表現型;在此類情況下需要進行酶活性測試以分配G6PD 表現型。 Tafenoquine的安全性已在G6PD酶活性≥正常的70%時確立 |
| 無法判定 | 攜帶至少一個等位基因具有功能尚未確定 | Dagua B/Dagua |
急性溶血性貧血風險未知 | 為確定G6PD狀態,必須測量酶活性。藥物使用應根據基於活性表現型的建議進行指導 | 中等建議 |
CNSHA: 慢性非球形紅細胞溶血性貧血
a WHO分類來自[文章:22293322],其他細節來自[文章:4963040]。I類等位基因s極為罕見;II類和III類等位基因s之間的區分不明確。幾乎所有患者都會攜帶II類、III類或IV類等位基因s
b 由於G6PD 等位基因s數量龐大,除了這裡給出的例子外,還可能存在其他基因型;請參見G6PD 等位基因定義表以獲得更全面的等位基因s列表和G6PD 等位基因功能表以了解其分配的功能(WHO類別)。注意某些實驗室使用“B 等位基因”來指示不攜帶已知I-III類變異的等位基因。G6PD頻率表可用於查閱主要生物地理群體中的G6PD 等位基因s頻率
c 評分方案在指引補充材料的建議強度部分中描述
d 此類基因型從未見過,推測極為罕見
e 由於X 染色體聯鎖嵌合現象(X-linked mosaicism),異型合子(Heterozygous)(通常為女性)攜帶一個非缺乏(IV類)和一個缺乏(I-III 等位基因s)等位基因可能顯示正常或缺乏 表現型。因此很難預測這些個體的表現型(見補充材料,G6PD異合子)
f
2018年9月更新
CPIC作者建議將G6PD A變異分類為IV/功能正常(先前為II-IV/缺乏-功能正常),基於支持功能的新證據[文章:27040960]和[文章:30206300]。此變更已納入G6PD 等位基因定義表
2014年8月
2014年5月在線接受文章預覽,2014年6月11日提前在線發表
- 關於使用藥物基因組學測試來確定是否應進行rasburicase治療的指引已由臨床藥物基因體學實施聯盟(CPIC)在《Clinical Pharmacology and Therapeutics》期刊中發表
- 2014年rasburicase指引摘錄:
- 「如上所述,FDA、EMA和PMDA在G6PD缺乏症患者中禁用rasburicase(32-34)(見表2)。如果基於基因分型可以明確地分配缺乏狀態,那將是使用rasburicase的充分禁忌。然而,由於基因測試的局限性(如上所述),在大多數情況下,必須進行G6PD酶測試以分配G6PD狀態。」
- 這些指引適用於
- 新生兒
- 兒科
- 成人
- 下載並閱讀:
表1:根據G6PD 表現型推薦的rasburicase劑量
改編自2014年指引的表1和表2
| 表現型(基因型)a | 雙倍型範例b | 對表型測量的影響 | rasburicase的劑量建議 | 建議等級 c |
|---|---|---|---|---|
| 正常d。攜帶非缺乏(IV類)等位基因的男性或攜帶兩個非缺乏(IV類)等位基因s的女性 | 男性:B, Sao Boria。女性:B/B, B/Sao Boria | 溶血性貧血風險低或降低 | 基於G6PD狀態,無理由停止rasburicase d | 強烈建議 |
| 缺乏或嚴重缺乏合併慢性非球形紅細胞溶血性貧血(Deficient with CNSHA)。攜帶I、II或III類等位基因的男性,攜帶兩個缺乏 I-III類等位基因s的女性 | 男性:A-, Orissa, Kalyan-Kerala, Mediterranean, Canton, Chatham, Bangkok, Villeurbanne。女性:A-/A-, A-/Orissa, Orissa/Kalyan-Kerala, Mediterranean/Mediterranean, Chatham/Mediterranean, Canton/Viangchan, Bangkok/Bangkok, Bangkok/Villeurbanne | 急性溶血性貧血風險 | rasburicase禁用;替代方案包括allopurinol e | 強烈建議 |
| 可變型(Variable) d,f。攜帶一個非缺乏(IV類)和一個缺乏(I-III變異)等位基因的女性 | B/A-, B/Mediterranean, B/Bangkok | 溶血性貧血風險未知 | 為確定G6PD狀態正常,必須測量酶活性;替代方案包括allopurinol e | 中等建議 |
a「類別」指的是WHO分類,來自[文章:22293322],其他細節來自[文章:4963040]。I類變異極為罕見;II類和III類變異之間的區分不明確;而「V類」非常高活性變異僅在一個案例中報導過[文章:4963040]。因此,幾乎所有患者都會攜帶II類、III類或IV類等位基因s。應注意,變異的類別可能僅根據患者的臨床表現分配,隨後識別出變異
(*) Luzzatto, L. & Poggi, V. 葡萄糖-6-磷酸脫氫酶缺乏症 在:Nathan和Oski的嬰幼兒和兒童血液學,第7版(編輯:Meloni, D., Anderson, A. 書籍作者:Orkin, S.H., Fisher, D.E., Look, A.T., Lux IV, S.E., Ginsburg, D., Nathan, D.G.)(Saunders, Elsevier., 2009)
b 由於G6PD變異數量龐大,除了這裡給出的例子外,還可能存在其他雙倍型;請參見補充表S1以獲得更全面的變異等位基因s列表及其分配的WHO類別
c 評分方案在補充材料中描述(見建議強度材料)
d 陰性或不確定的基因測試不能被認為表示正常的G6PD 表現型;在此類情況下需要進行酶活性測試以分配G6PD 表現型
e allopurinol與攜帶HLA-B*58:01 等位基因的罕見攜帶者的嚴重皮膚反應相關[文章:23232549]
f 由於X 染色體聯鎖嵌合現象(X-linked mosaicism),女性異型合子(Heterozygous)攜帶一個非缺乏(IV類)和一個缺乏(I-III變異)等位基因可能顯示正常或缺乏 表現型。因此很難預測這些個體的表現型(補充材料,G6PD異合子)
圖1:解釋G6PD基因型和評估是否需要酶活性測試的工作流程
圖1來自指引
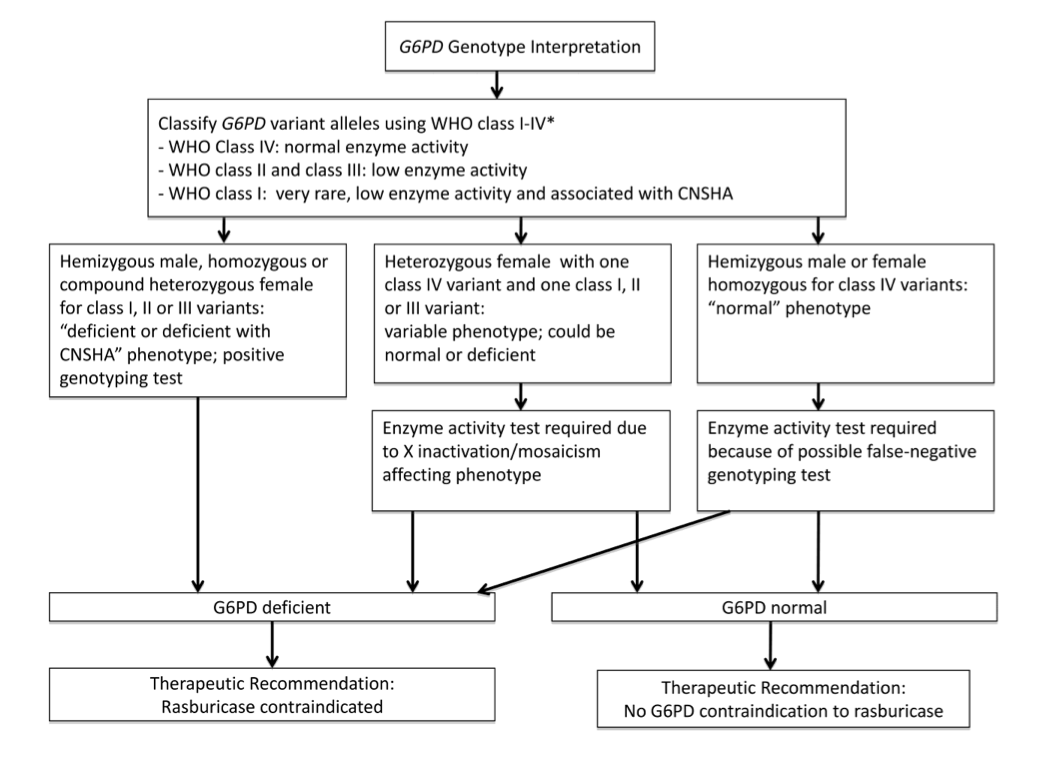
*應注意,變異的類別可能僅根據患者的臨床表現分配,隨後識別出變異[文章:22293322]
Uric Acid-Lowering Drugs 藥效學途徑
概括
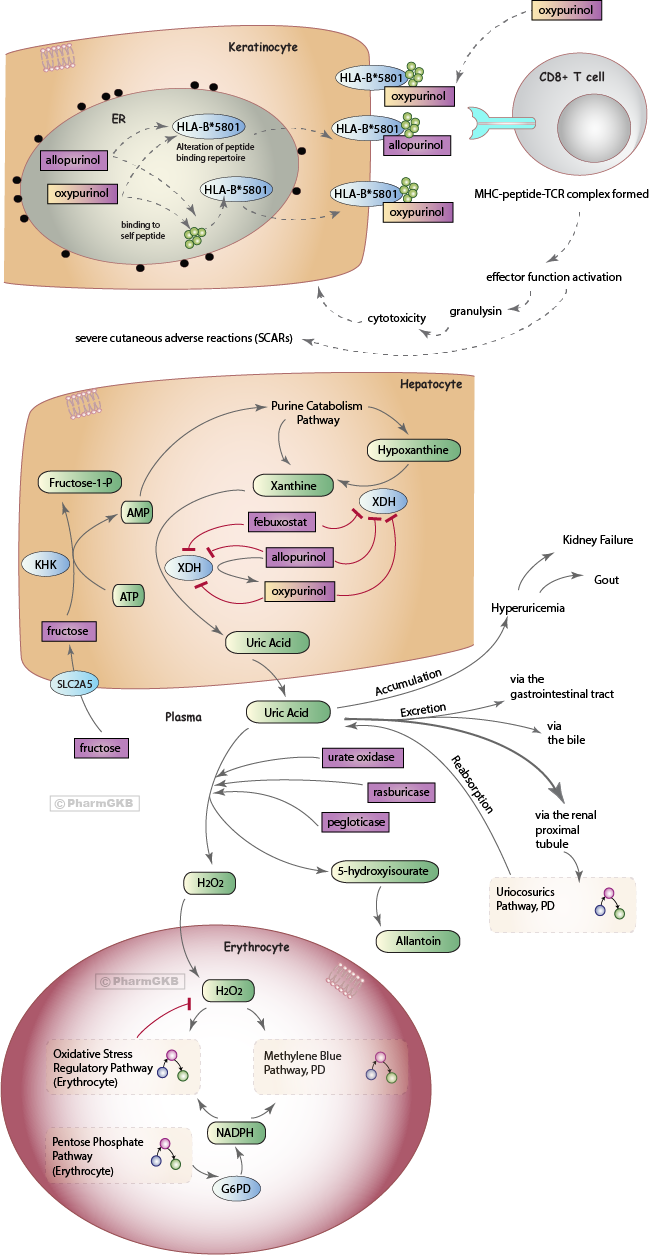
示意圖顯示可抑制尿酸形成或促進其排泄之藥物,以及與這些藥物相關的不良反應。
描述
背景
人類的尿酸生成
嘌呤對細胞的生長和存活至關重要;作為DNA和RNA的組成部分,作為代謝反應的輔酶,以及細胞訊息傳遞途徑的組成部分[Article:11934348]。在人體和大型猿類中,尿酸是嘌呤CAT代謝的最終產物途徑;較低的生物體表達尿酸氧化酶,進一步降解尿酸,但由於尿酸氧化酶基因中的無義突變(UOX pseudogene),在人體中不表達[Articles:12646938,1765273,11934348](Keilin 1959)。在生理pH下,尿酸主要以尿酸陰離子的形式存在[Article:20613716]。在此,我們將尿酸統稱為「尿酸」。尿酸主要通過腎臟清除(>70%),少量通過膽汁和腸道分泌[Articles:11934348,22665944,22359229,20613716,22038265]。然而,約90-95%的尿酸被近端腎小管中的轉運蛋白重吸收[Articles:20613716,22359229]。因此,血漿尿酸的濃度取決於幾個因素;尿酸的生成和排泄、嘌呤de novo合成、CAT代謝和周轉[Article:11934348]。
高尿酸血症
血漿尿酸水平超過約7-8mg/dL定義為「高尿酸血症」;這是一種由於尿酸的生成或釋放增加,超過正常腎臟清除能力,或腎臟清除減少或尿酸重吸收增加的情況所引起的[Articles:16597166,18838473,22359229](Fojo, 2011)。高尿酸血症的一個主要風險是由於尿酸在腎小管酸性環境中的結晶化而導致腎功能衰竭[Article:12646938]。因此,慢性腎病與高尿酸血症TEN相關[Articles:20562597,12646938,22665944]。持續的高尿酸血症可能導致痛風(或痛風性關節炎),當單鈉尿酸晶體在關節中釋放到關節腔中並引起炎症時。痛風是一種使人衰弱的疾病,與更高的共病風險和更高的死亡率相關[Articles:20562597,22069122,22198943,22945592]。然而,並非所有ALL未合併的高尿酸血症患者都會發展為痛風,因此不建議進行藥物治療[Article:17349440]。高尿酸水平也與高TEN血壓、肥胖和胰島素抵抗的發展相關[Articles:19151107,22359229]。高尿酸血症在癌症患者和接受化療的患者中常見,因為惡性細胞產生更多的嘌呤,並通過細胞周轉增加或化療引起的細胞溶解釋放細胞成分(也稱為腫瘤溶解綜合徵,TLS)(Fojo, 2011) [Articles:12646938,11934348,18838473]。
(非PMID參考資料:Fojo AT: Metabolic Emergencies. In DeVita, Hellman, and Rosenberg's Cancer: Principles & Practice of Oncology, 9th Edition. Volume 9th Edition. Edited by DeVita VTL, Theodore S.; Rosenberg, Steven A.: Lippincott Williams & Wilkins; 2011)。
飲食
飲食可能會影響血漿尿酸水平。酒精和飲食中的嘌呤(肉類、海鮮)是痛風的可能風險因素,歷史上被視為富人的疾病[Articles:22645377,23253231,23175570]。啤酒中含有高量的鳥苷,乙醇增加ATP的降解[Articles:23253231,23175570]。糖(蔗糖 sucrose)是一種由葡萄糖和fructose組成的二糖[Article:23175570]。當葡萄糖被代謝時,細胞內ATP的水平會保持,但對於fructose則不然[Article:23175570]。fructose通過SLC2A5(Glut5)運輸進入細胞,並由酮己糖激酶代謝為fructose-1-磷酸(KHK),該酶利用ATP [Articles:24065788,23175570,18398011]。這主要發生在肝臟中,導致細胞內ATP和磷酸鹽水平的瞬時降低[Articles:24065788,23175570]。隨後,腺苷單磷酸(AMP)去胺酶被激活,AMP由fructose的代謝產生,進入嘌呤CAT代謝途徑,最終導致尿酸作為最終產物[Articles:24065788,23175570]。假設是,最近在不同國家糖(和fructose)消費的增加與痛風、代謝疾病和糖尿病的增加相關,並且部分可歸因於尿酸水平的升高[Articles:23175570,19151107,22198943,23493538]。然而,fructose的攝入是否會導致臨床上顯著的尿酸水平升高,並隨之發展為代謝疾病或痛風,仍然是一個有爭議的話題,是否應建議患者減少fructose的攝入以降低尿酸水平仍然是爭論的焦點 - 有關更多細節,請參見其他文章:[Articles:23175570,24065788,23793849,22645377,22457397,18163396,21889564]。據我們所知,目前沒有針對fructose的攝取或代謝的藥物用於治療高尿酸血症。
藥效學
治療痛風或高尿酸血症的主要藥理策略有三種:(1)減少尿酸的生成,(2)增加尿酸的排除,(3)減少尿酸的重吸收。這途徑討論了allopurinol和febuxostat通過第一種機制起作用的藥物,以及通過第二種機制起作用的尿酸氧化酶。它們對尿酸途徑的作用機制如上圖所示,並在下文中描述。與這些藥物反應相關的遺傳變異詳述。通過第三種機制在腎臟中起作用的尿酸排泄劑類藥物在促尿酸排泄劑(Uricosurics)藥效學途徑中進行討論。
allopurinol
allopurinol是一種指示用於治療痛風、化療患者高尿酸血症的預防以及預防腎結石復發的藥物(藥物仿單)。allopurinol及其代謝物氧嘌呤醇是次黃嘌呤和黃嘌呤的類似物,通過與黃嘌呤脫氫酶結合並抑制其活性來防止尿酸的形成(見上圖)[Articles:20562597,12646938,16597166,22665944]。次黃嘌呤和黃嘌呤通過尿液排出或在核苷酸和核酸的合成中再利用[Article:11934348]。allopurinol也可能具有有益的止痛效果,可能是通過增加次黃嘌呤產生的腺苷(這些效果會被咖啡因抑制)[Articles:19133997,19133987]。然而,allopurinol治療的一個缺點是它不清除現有的高血漿尿酸水平,並且由於黃嘌呤的溶解度低於尿酸,可能導致黃嘌呤腎結石或黃嘌呤腎病[Articles:11934348,12646938]。
allopurinol可能會改變與之同時服用的藥物的反應。前藥硫唑嘌呤和6-巰基嘌呤通過次黃嘌呤磷酸核糖轉移酶1(HPRT1)轉化為活性代謝物,或被XDH及其他酶失活(見硫嘌呤途徑,PK/PD)[Article:22132961]。將allopurinol與硫嘌呤療法聯合使用可增強HPRT1的活性,並與活性硫嘌呤代謝物的顯著增加相關[Article:22132961]。allopurinol降低甲氨蝶呤的清除率,這與口腔黏膜炎的風險增加相關[Article:19834958]。
febuxostat
febuxostat(Uloric®)是一種非嘌呤抑制劑,能夠結合並抑制尿酸氧化酶的氧化或還原形式,並不EEM與嘌呤或嘧啶CAT代謝相互作用途徑(見圖)[Articles:20562597,19247302,20401506]。它被指示用於治療痛風患者的高尿酸血症,但不適用於無症狀的高尿酸血症。它是對於因allopurinol因過敏反應而被禁用的患者的替代療法[Articles:22582566,20401506,19247302],並且在多項美國研究中顯示出80mg的療效優於標準allopurinol治療,且不良事件發生率相當[Articles:22316106,22436129,20370912,23929928],儘管allopurinol的療效率與歐洲人不同[Article:20401506]。febuxostat在接受硫唑嘌呤或巰基嘌呤治療的患者中是禁忌的,因為藥物交互作用。
尿酸氧化酶(Uricase)
增強尿酸排泄的一種策略是添加外源性尿酸氧化酶,這在人體中不表達。尿酸氧化酶將尿酸分解為5-羥基異尿酸,然後在不需要酶的情況下自發降解為ALL酸(見圖)[Articles:12646938,16597166,20562597]。ALL酸的溶解度比尿酸高5-10倍,因此更容易通過腎臟排泄[Articles:11934348,16597166,12646938]。Uricozyme®是從Aspergillus flavus中提取的尿酸氧化酶,但由於產品相關的雜質,與急性過敏反應相關[Articles:11934348,20562597]。一種重組形式的尿酸氧化酶rasburicase(商業名稱為Elitek®、Fasturtec®、Rasuritek®)被開發以減少這些反應的發生[Articles:11934348,20562597]。rasburicase已被證明是一種比allopurinol更有效的治療,能迅速降低尿酸血漿濃度和暴露[Articles:9369411,11157020,11342423,11694947,20562597]。然而,rasburicase不適合用於治療痛風,因為其半衰期短,因此pegloticase(或PEG尿酸氧化酶,Krystexxa®)是重組尿酸氧化酶與聚乙二醇的結合,據認為與rasburicase相比,能降低免疫原性並延長半衰期[Articles:20562597,22198943]。最近對於健康血液供體中發現的抗PEG抗體的發展引起了關注(可能由於對此類化合物的接觸增加),這可能影響PEG乙基化藥物的療效[Article:22931049]。
藥物基因體學
HLA基因和allopurinol誘導的過敏反應
接受allopurinol的患者中,有一小部分(0.1%至0.4%)發展為危及生命的嚴重皮膚不良反應(SCARs),包括藥物過敏綜合徵(DRESS),史蒂芬-強森症候群及毒性表皮壞死溶解症(SJS/TEN)。allopurinol過敏反應與藥物的預期藥效途徑完全不同:SJS和TEN被認為是通過藥物特異性、HLA的I類限制性和顆粒溶素介導的途徑發生的[Articles:17620823,19029983]。NK細胞也可能參與其中[Article:19029983]。
HLA-B*5801 等位基因目前是與allopurinol誘導的SCAR最為確立的遺傳關聯,並已在多個人群中證實(表1)。人類HLA-B基因是基因組中多態性最豐富的基因,擁有超過1500個等位基因,由許多變異組成[Article:17620823]。歷史上,這些等位基因是通過抗原結合測定法識別的,但最近則利用測序和基因型技術(見表1)。標記HLA-B*5801 等位基因的單核苷酸多態性(SNPs)在不同人群中有所不同[Article:16998491]。HLA-B*5801特異性allopurinol誘導的過敏反應的機制尚未完全理解,但對於HLA-依賴的T細胞刺激的幾個假設可能適用[Article:22017685]。主要組織相容性複合體(MHC)I類分子(由HLA-A編碼,-B和-C)由大多數有核細胞表達,並參與將細胞內自我或外源肽呈遞給CD8+細胞毒性T細胞[Articles:22017685,17620823,22943588,23232549]。當呈遞自我肽(肽)時,與特定T細胞受體(TCRs)的相互作用較弱。然而,當呈遞外源/新肽時,會引發抗原特異性CD8+ T細胞反應,導致T細胞增殖和效應功能的激活(例如對呈遞細胞的細胞毒性)[Article:22017685]。allopurinol或氧嘌呤醇可能與自我蛋白/肽結合,形成一種經過抗原處理並由HLA-B*5801特異性呈遞的產品,以激活抗原特異性T細胞(hapTEN概念)[Article:22017685]。有假設認為,在低濃度下,信號不足以激活T細胞,但在較高劑量或由於腎功能衰竭(這兩者都是allopurinol誘導的潛在風險因素,見表1),藥物的濃度增加會導致T細胞的激活[Article:22943588]。allopurinol/氧嘌呤醇可能與HLA-B*5801-MHC-肽複合物直接在細胞表面相互作用,而不是經過抗原處理(pi概念)[Articles:22017685,24591375]。在內質網中HLA-B*5801的折疊過程中,allopurinol/氧嘌呤醇可能會被納入肽結合槽,潛在地改變HLA-B*5801能夠呈遞的自我肽的範圍,導致由於呈遞新肽而引起的ALL過敏反應(錨位修飾/佔據模型)[Article:22017685]。最近的一項體外研究提供了pi概念的證據,並且氧嘌呤醇在對HLA-*5801分子的親和力上高於allopurinol的對接實驗[Article:24591375]。
一項綜合分析計算了allopurinol誘導的SJS/TEN在HLA-B*5801攜帶者中的比值比為96.6,與allopurinol耐受的匹配對照相比,或與人群對照相比為79[Article:21906289]。美國風濕病學會(ACR)的指導方針建議僅篩查高風險人群(如3期慢性腎病的韓國人、漢族和泰國患者),並且那些被發現對HLA-B*5801陽性的人應該被處方替代藥物[Article:23024028]。藥物基因體學臨床應用聯盟(CPIC)的指導方針還建議不使用allopurinol於已知攜帶HLA-B*5801的患者[Article:23232549]。目前批准的FDA-藥物仿單對allopurinol的基因型沒有任何信息,儘管FDA的科學家發表了一項研究,評估allopurinol的臨床實用性,並報告了強烈建議和高度顯著的HLA-B*5801與allopurinol相關的可能性之間的關聯[Article:22118056]。
在某些人群(漢族、泰國、韓國)中,HLA-B*5801解釋了80-100%的SCAR病例,而在其他人群(日本、歐洲)中,HLA-B*5801解釋了約55%的病例[Articles:15743917,18192896,19018717,19696695,21301380,21545408,21912425],這表明還有其他風險等位基因、遺傳變異和其他因素對SCAR也很重要。體外實驗也表明,allopurinol/氧嘌呤醇誘導的反應並不僅限於HLA-B*5801分子[Article:24591375。與allopurinol誘導的SCAR相關的其他等位基因包括:rs9263726 A(在日本患者中與HLA-B*5801完全連鎖),rs2734583,rs3094011,rs2844665 C,rs3815087 A,rs3130931 C,rs3130501 G,rs3094188 A,rs9469003 C,HLA-A*33:03,HLA-C*03:02,HLA-C*08:01,HLA-Cw3,HLA-A33,HLA-DR13,HLA-D3,HLA-Cw*0302,HLA-A*3303,HLA-DRB1*0301,rs3117583,rs1150793,rs2855804,rs2268791,rs1594,rs2304224(每項研究的詳細信息可在allopurinol PGx研究頁面中找到)[Articles:21912425,21801394,21545408,21301380]。
febuxostat與兩例先前對allopurinol有不良反應的患者相關的過敏反應[Articles:22383358,21724706]-據我們所知,這方面的遺傳機制尚未描述。目前未找到與藥物基因體學相關的研究。
+表1:報告的HLA-B*58:01與allopurinol誘導的SCAR風險之間的關聯。+
基於序列的分型。| 研究類型和參考 | 病例組 | 對照組 | OR(95% C.I.) | P值 | 患者人群 | 種族/民族 | 分型和基因分型方法 |
|---|---|---|---|---|---|---|---|
| Case-control study [Article:23669020] | n=7(SJS,SJS眼型,紅斑性多形性紅斑(EEM)輕型)。 | n=25(無ADR)。 | 65.6(2.9-1497) | 9.733x10^-4 | 高尿酸血症。 | 日本人。 | PCR-rSSO和PCR-SBT. |
| Case-control study [Article:23600531] | n=25(SJS/TEN,DRESS)。 | n=23 allopurinol-耐受。 | 39.11(4.49-340.51) | 5.9x10^-4 | 無症狀高尿酸血症、痛風性關節炎或腎病。 | 葡萄牙人。 | PCR-rSSO和SSO-HR分型試劑盒。 |
| 家庭成員的觀察性研究 [Article:23280169] | n=1名男性,發展為SJS。 | n=1名病例的兄弟,allopurinol-耐受。 | NA | NA | 痛風和原發性高TEN血壓。 | 自我描述的漢族。 | 4位數,高分辨率DNA測序。 |
| Case-control study [Article:22348415] | n=20(SJS,DRESS,TEN,EMM)。 | n=30 allopurinol-耐受,至少治療1年(未與病例匹配)。 | 123.5(12.8-1195.1)(OR在排除EMM病例時更高)。 | <1x10^-4 | NA | 漢族(香港)。 | |
| Case-control study [Article:15743917] | n=51名患者,因allopurinol誘導的SCAR(SJS、TEN或HSS)。 | n=135 allopurinol-耐受(至少6個月的治療)。 | 580.3(34.4-9780.9) | 4.7 x10^-24 | 高尿酸血症。病例與對照相比,慢性腎功能不全的發生率顯著較高,而痛風性關節炎則相反。 | 居住在台灣的漢族。 | 基於序列特異性寡核苷酸的反向線條印跡。 |
| 比較等位基因在一個人群中的頻率 [Article:18192896] | n=31名患者,因allopurinol誘導的SJC或TEN。 | NA | 61(32-118)為病例的等位基因頻率,與一般歐洲人群中的等位基因頻率頻率相比。 | <10^-8 | 主要是高尿酸血症。 | 混合人群,主要是歐洲人。 | OLERSUP SSP HLA-B試劑盒,三例中的測序。 |
| 比較等位基因在一個人群中的頻率 [Article:19018717] | n=10名患者,因allopurinol誘導的SJS,TEN(一名患者同時接受carbamazepine治療,另一名患者同時接受phenytoin治療)。 | NA | 40.83(10.53-158.9)等位基因頻率與493名健康日本受試者的頻率相比。 | <0.0001 | NA | 日本人。 | 測序。 |
| Case-control study [Article:19696695] | n=27名患者,因allopurinol誘導的SJS或TEN(在治療的前三個月內)。 | n=54名allopurinol-耐受患者(>6個月治療)來自同一家醫院。 | 348.3(19.2-6336.9) | 1.6x10^-13 | 高尿酸血症,部分患者伴隨痛風性關節炎。 | 自我認定的泰國或泰國華人。 | PCR使用序列特異性引物和基於序列的分型。 |
| Case-control study [Article:21301380] | n=25名患者,因allopurinol誘導的SCAR(20名患者有DIHS,5名患者有SJS/TEN)。 | n=57名allopurinol-耐受患者。 | 97.8(18.3-521.5)(病例與耐受對照)。 | 2.45 x10^-11 | 病例=與慢性腎功能衰竭相關的痛風性關節炎患者。對照=慢性腎功能衰竭患者。 | 韓國人。 | 直接DNA測序分析。 |
| 比較等位基因頻率在健康個體中的頻率 [Article:21545408] | n=7名allopurinol-誘導的SJS/TEN患者。 | n=115名健康個體。 | 13.625(2.774-69.448) | 0.248經多重比較修正後。 | NA | 白人,意大利北部。 | 4位數等位基因水平在抗原結合域內。PCR-SSP |
| Case-control study [Article:21393610] | n=16名患者,出現allopurinol過敏反應(9名有SCAR,7名有簡單皮疹)。 | n=432名allopurinol-耐受患者(60天)。 | 179.24(10.19-3151.74)SCAR患者與耐受者。 | <0.001 | 慢性腎功能不全患者服用allopurinol。 | 韓國人。 | 微淋巴細胞毒性法進行的血清學HLA分型以檢測HLA-B*58。 |
| Case-control study [Article:22909208] | n=38名allopurinol-誘導的MPE,DRESS,SJS/TEN(在接觸的前兩個月內)。 | n=63名allopurinol-耐受(治療>3個月且無皮膚表現)。 | 580.07(32.18-10456.80) | 7.01 x10^-18 | 高尿酸血症和痛風性關節炎。(病例中觀察到慢性腎功能不全的發生率較高)。 | 病例=來自中國南部地區,對照=ALL漢族。 | 直接DNA測序。 |
| Case report [Article:19483528] | n=1,一名8歲女孩在TEN治療後10天被診斷。 | NA | NA。檢測顯示她有HLA-B*5801。 | NA | 因抗結核治療而發展為無症狀高尿酸血症,並接受allopurinol治療。 | 德國、肯尼亞父母。 | 未描述。 |
| Case report [Article:22901319] | n=1,一名65歲男性在DRESS治療開始後1個月發展。 | NA | NA。檢測顯示他有HLA-B*5801陽性基因型。 | NA | 高尿酸血症。 | 漢族。 | 方法未描述。 |
| 該研究確定了澳洲各地有allopurinol hypersensitivity的患者,並進行了基因分型。[Article:21790926] | N=11名患者,具有allopurinol過敏性,包括SJS/TEN和DRESS/DIHS,N=12名患者具有MPE。 | NA | NA。HLA-B*5801在6/5例具有SJS/TEN中發現,5/5例具有DRESS/DIHS中未發現。 | NA | 未描述。 | 澳大利亞,混合人群:白人和東南亞人。 | 四位數高分辨率DNA基於序列的HLA分型。 |
| Case report [Article:17587850] | n=1,一名57歲男性在SJS治療後10天被診斷。 | NA | NA。分型顯示他有HLA-A31、A33、B51和B58。 | NA | 未描述。 | 未描述。 | 反向序列特異性寡核苷酸與PCR進行血清學HLA分型。 |
| Case report [Article:17587850] | n=1,一名77歲男性被診斷為allopurinol誘導的DIHS。 | NA | NA。分型顯示他有HLA-A31、A33、B39和B58。 | NA | 未描述。 | 未描述。 | 未描述。 |
| Case report [Article:17587850] | n=1,一名93歲男性被診斷為allopurinol誘導的SJS/TEN(一年前他曾在allopurinol治療後1個月內出現過紅斑性丘疹和發燒)。 | NA | NA。他有HLA-A24、A33和B52。 | NA | 未描述。 | 未描述。 | 未描述。 |
| 藥物監測計劃評估allopurinol皮膚不良反應[Article:22017528] | n=84例,包括紅斑丘疹、SJS、TEN和DRESS。對於HLA-B*5801的檢測僅在一小部分有SJS/TEN的患者中進行。 | 等位基因頻率在一般歐洲人群中的頻率為0.015進行比較。 | 18/18例SJS/TEN中進行檢測的患者均攜帶HLA-B*5801。OR=65.07(30.66-138.09),與一般歐洲人群相比等位基因頻率 | <0.0000。 | 無症狀高尿酸血症、痛風或繼發性高尿酸血症(許多患者也有高TEN心臟病和腎功能衰竭)。 | 南薩丁尼亞,意大利,歐洲。 | PCR-SSO/ PCR-SSP。 |
尿酸氧化酶和參與氧化壓力調節的基因
rasburicase和pegloticase在G6PD 缺乏個體中是禁忌的,因為增加了溶血性貧血和高鐵血紅蛋白症的風險,這些情況涉及紅血球(RBCs)(鏈接到藥物仿單)[Article:15862084]。當rasburicase將尿酸分解為ALL酸時,會釋放出過氧化氫(H2O2)作為副產物,這是一種反應性氧物種,可能對RBCs造成損害,最終導致細胞周轉(溶血)。在正常情況下,H2O2通過調節機制被還原為水和氧分子,其中許多需要NADPH(見氧化壓力調節途徑)[Articles:23913015,16597166,12646938,15862084,8704218,2633878,18177777。G6PD是五碳磷酸途徑(PPP)中的一種酶,生成NADPH。這途徑是RBCs中NADPH的唯一來源(見五碳磷酸途徑)[Articles:18177777,2633878,4154443]。G6PD 缺乏 RBCs無法增強PPP產生的能力,因此更容易受到氧化損傷,最終導致高鐵血紅蛋白症和/或溶血[Articles:4154443,2633878,15862084,22024786,7073040,15862084,10533013]。迄今為止,已描述了超過180種基因變異,這些變異在G6PD基因中,許多導致RBCs中G6PD酶的缺乏[Articles:22293322,17611006,5316621,2633878]。