CPIC 指南註解:tegafur 與 DPYD 基因
摘要
截至2017年11月更新的CPIC指南中關於fluoropyrimidine劑量的建議,已不再根據DPYD基因型對tegafur進行劑量建議。這是由於關於DPYD變異對tegafur毒性風險影響的證據有限。目前,該指南僅適用於5-fluorouracil和capecitabine的劑量建議。
註釋
此註釋基於CPIC® 關於fluoropyrimidines和DPYD的指引。
2017年11月更新
2017年11月提前在線發表
- 關於5-fluorouracil和capecitabine的2017年CPIC指引更新已被接受發表於《Clinical Pharmacology and Therapeutics》期刊。審查了截至2017年3月的文獻,並更新了建議和補充信息。特別是,劑量建議已修改為僅適用於5-fluorouracil和capecitabine;不再適用於tegafur。這是由於關於DPYD變異對tegafur毒性風險影響的證據有限。
- 下載並閱讀:
2014年5月更新
- CPIC作者建議將DPYD*4、*5、*6和*9A等位基因s分類為「正常」活性,部分基於最近發表的DPYD變異的潛在臨床相關性對二氫嘧啶脫氫酶活性的比較功能分析。
2013年12月發表
2013年8月在線接受文章預覽;2013年10月提前在線發表。
- 關於在fluoropyrimidines劑量中使用藥物基因組學測試的指引已由臨床藥物基因體學實施聯盟(CPIC)發表於《Clinical Pharmacology and Therapeutics》期刊。
- 這些指引適用於:
- 在撰寫本文時,尚無關於DPYD*2A、*13或rs67376798在兒科患者群體中對5-fluorouracil毒性的可能作用的數據;然而,沒有理由懷疑DPYD變異等位基因s會在兒童中與成人不同地影響5-fluorouracil的代謝。
- 基於DPYD基因型的fluoropyrimidine劑量指引摘錄:
- 「劑量建議的強度基於某些變異(DPYD*2A、*13和rs67376798)明顯影響DPD活性,而DPD活性明顯與5-fluorouracil清除率相關,5-fluorouracil暴露與其毒性效應相關。因此,對於具有這些變異的患者,減少fluoropyrimidine劑量可能預防嚴重甚至危及生命的毒性。然而,現有證據並未明確指出需要減少多少劑量以預防fluoropyrimidine相關毒性...[基於文獻回顧(見完整手稿),]我們的建議是從至少減少起始劑量的50%開始,然後在未經歷或臨床可耐受毒性的患者中增加劑量以維持療效,在不耐受起始劑量的患者中減少劑量以最小化毒性,或進行藥代動力學指導的劑量調整(如果可用)。對於同型合子(Homozygous)為DPYD*2A、*13或rs67376798的患者,可能表現出完全的DPD缺乏,不建議在這些患者中使用5-fluorouracil或capecitabine。」
- 下載並閱讀:
表1:根據基因型/表現型推薦的fluoropyrimidines劑量
改編自2013年指引手稿的表1和表2。
| 表現型(基因型) | 雙倍型的例子 | 對表型測量的影響 | 劑量建議 | 建議等級 a |
|---|---|---|---|---|
| 同型合子(Homozygous)野生型或正常,高DPD活性(兩個或更多功能性*1 等位基因s) | *1/*1 | 正常DPD活性和「正常」fluoropyrimidine毒性風險 | 使用仿單推薦的劑量和給藥 | 中等建議 |
| 異型合子(Heterozygous)或中等活性(約3-5%的患者),可能有部分DPD缺乏,藥物暴露時有毒性風險(一個功能性等位基因 *1,加上一個非功能性等位基因 - *2A、*13或rs67376798A c) | *1/*2A; *1/*13; *1/ rs67376798A c) | DPD活性降低(白細胞DPD活性為正常人群的30%至70%)且使用fluoropyrimidine藥物治療時增加嚴重甚至致命藥物毒性的風險 | 從至少減少起始劑量的50%開始,然後根據毒性b或藥代動力學測試(如果可用)進行劑量滴定 | 中等建議 |
| 同型合子(Homozygous)變異,DPD缺乏(約0.2%的患者),藥物暴露時有毒性風險(2個非功能性等位基因s - *2A、*13或rs67376798A c) | *2A/*2A; *13/*13; rs67376798A c / rs67376798A c | 完全DPD缺乏且使用fluoropyrimidine藥物治療時增加嚴重甚至致命藥物毒性的風險 | 選擇替代藥物 | 強烈建議 |
a 評分方案描述於2013年補充中。
b 在未經歷或臨床可耐受毒性的患者中增加劑量以維持療效;在不耐受起始劑量的患者中減少劑量以最小化毒性。
c 注意rs67376798A 等位基因指的是正向染色體鏈上的等位基因。這很重要,因為DPYD位於負向染色體鏈上,rs67376798是一個T/A snp。因此,基因上的T 等位基因賦予缺乏,而正向染色體鏈上的互補(A 等位基因)則表示缺乏。
Fluoropyrimidine 途徑, 藥效學
概括
非組織特異性癌細胞模型,顯示可能參與 fluoropyrimidines(5-fluorouracil(5-FU)、capecitabine、tegafur)藥效學之基因
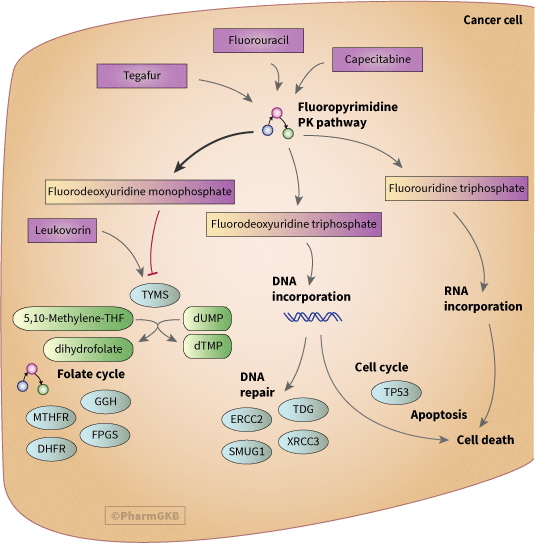
描述
Fluoropyrimidine 是一類廣泛用於治療癌症的抗代謝藥物,包括結直腸癌、乳腺癌及呼吸消化道癌症。對於作為一線單藥治療的 Fluoropyrimidine 5-fluorouracil (5-FU) 的反應率較低(對於晚期結直腸癌為 10-15%),因此通常與其他細胞毒性藥物如奧沙利鉑(稱為 FOLFOX)和伊立替康 (FOLFIRI) 一同使用。這樣可以提高藥物的療效,對於晚期結直腸癌的反應率可達 40-50%。由於這些常見的聯合治療,對 5FU 的體內藥物基因組學效應的研究可能會變得複雜。通過表達研究,已發現與 Fluoropyrimidine 相關的候選基因,這些研究通過體外處理細胞來識別藥物抗性和敏感性的標記,儘管這些大多數尚未在患者研究中得到驗證 [Articles:16510598, 19219653, 19339911, 15548681, 16709241, 19302291].
Fluoropyrimidine 的主要作用機制被認為是抑制胸苷酸合成酶 (TYMS),但最近的證據也顯示出其他藥效學 途徑 作用,通過將藥物代謝物納入 DNA 和 RNA 中。Fluoropyrimidine 被分解為三種具有藥效學作用的代謝物,分別是 fluorodeoxyuridine monophosphate (FdUMP)、氟脫氧尿苷三磷酸 (FdUTP) 和 fluorouridine triphosphate (FUTP)(詳情請參見 PK 途徑)。在臨床上,5FU 通常以與 leucovorin (LV; 5-形式四氫葉酸) 的靜脈推注或持續輸注的方式給予。5FU 的作用機制可能因給藥方式的不同而有所不同,靜脈推注治療更有利於 RNA 損傷,而持續治療則更有利於 DNA 損傷 [Articles:19383847, 8996164].
FdUMP 與 TYMS 形成共價複合物 [Article:15638735],並阻止 dUMP 轉化為 dTMP,這對於嘧啶和 DNA 的合成是必要的,並阻止 5, 10-亞甲基四氫葉酸同時轉化為二氫葉酸,這是葉酸 途徑 的關鍵成分,能回收甲基並合成蛋氨酸。TYMS 的抑制導致 dUTP 和 dTTP 的不平衡,並增加 dUTP 錯誤納入 DNA 的情況 [Article:19402749]。FdUMP 與 TYMS 的複合物通過聯合給予可以替代 5,10-亞甲基 THF 的葉酸類似物來穩定,例如 LV (5-形式四氫葉酸) [Article:15638735]。該蛋白質以二聚體形式運作,兩個亞基均可結合核苷酸和葉酸。雖然已生成多種 TYMS 的結構,但許多是非人類蛋白質,且尚未有顯示人類 TYMS 與 FdUMP 結晶的結構可用 (PDB:1TSN 顯示 FdUMP 與大腸桿菌 TYMS 的結合)。
由於其參與內源性葉酸的代謝,LV、葉酸的給予及其他葉酸循環酶的活性可能會影響 TYMS 的活性。GGH 和 FPGS 的表達影響人類結腸 癌細胞 中還原葉酸的水平,從而決定 LV 對 5FU 細胞毒性的增強 [Article:18035049]。已顯示 DHFR 的表達在腫瘤細胞中與正常細胞相比有所改變 [Article:15814641],儘管它並不是 Fluoropyrimidine 的直接靶點,如同對甲氨蝶呤的情況,但它可能通過改變葉酸的可用性來影響 Fluoropyrimidine 的藥效學。腫瘤的 表現型 也可能在葉酸和甲基化方面對此 途徑 具有重要性。例如,結直腸癌的 CpG 島甲基化酶 表現型 (CIMP+) 中,基因啟動子超甲基化的情況已與 5FU 基礎治療的良好預後相關 [Article:19093176].
對於 DNA 由 dUTP 或 FdUTP 納入 DNA 所造成的損傷是否是 Fluoropyrimidine 的細胞毒性原因仍存在一些爭議 [Article:19402749]。無論是尿嘧啶還是 5FU 納入 DNA,所產生的損傷都是由於增加的碱基切除修復導致 DNA 斷裂,最終導致細胞死亡。SMUG1,一種尿嘧啶-DNA 糖苷酶,能從 DNA 中切除 5FU,並在體外保護細胞免受死亡 [Article:17283124]。最近的一篇論文提供了體外證據,顯示胸苷 DNA 糖苷酶 TDG 是負責 5FU 切除相關 DNA 斷裂的主要碱基切除酶 [Article:19402749].
TYMS 變異與 TYMS 的表達及對 fluorouracil 化療的反應有關,詳見於 [Articles:16267625, 17716232](見 TYMS VIP 註解以獲取更多細節)。儘管有許多研究檢查這些變異的影響,但來自異質性研究的矛盾發現使得尚未為臨床使用開發出明確的預測策略。最近一項對結直腸腫瘤樣本中 拷貝 數量變異的研究顯示,高 拷貝 數量與疾病復發和死亡相關 [Article:18607850],這表明簡單的基因分型可能無法提供完整的圖景。關於 MTHFR 變異對 Fluoropyrimidine 藥效學的影響存在矛盾數據(有關這些變異的更多細節,請參見 MTHFR VIP 註解)。最近的一篇綜述 [Article:19144510] 討論了 MTHFR 變異對腫瘤反應、疾病進展、生存和毒性的影響,指出許多顯示這些變異無影響的研究涉及與其他抗腫瘤藥物的聯合治療,而顯示相關性的研究則涉及單獨使用 Fluoropyrimidine 或與 LV 的治療。他們及其他人建議基於 途徑 的多變量方法可能是預測 Fluoropyrimidine 藥物反應的最有效方法 [Articles:19144510, 16785472, 15814641]。還有一些額外的出版物將變異與 DNA 修復酶和細胞週期 途徑 相關聯,涉及 藥物基因體學 中的 Fluoropyrimidine 在體內的作用 [Articles:17549067, 18267032, 18357466]。雖然在圖示中與細胞週期和凋亡的 途徑 部分一起呈現,但這個 TP53 變異可能通過調節 MTHFR 和 DHFR 來發揮作用,如在一項對接受 5FU 和美托黴素治療的乳腺癌患者的研究中所示 [Article:18498133].
總結來說,雖然 Fluoropyrimidine 在臨床上已使用超過 50 年,且藥效學 途徑 中的幾個候選基因已顯示影響臨床結果,但許多數據是矛盾的,並且因聯合治療方案而變得複雜。因此,尚未證明出一個明確的預後或預測測試策略。
Fluoropyrimidine 途徑, 藥物動力學
概括
fluoropyrimidines 代謝途徑之示意圖
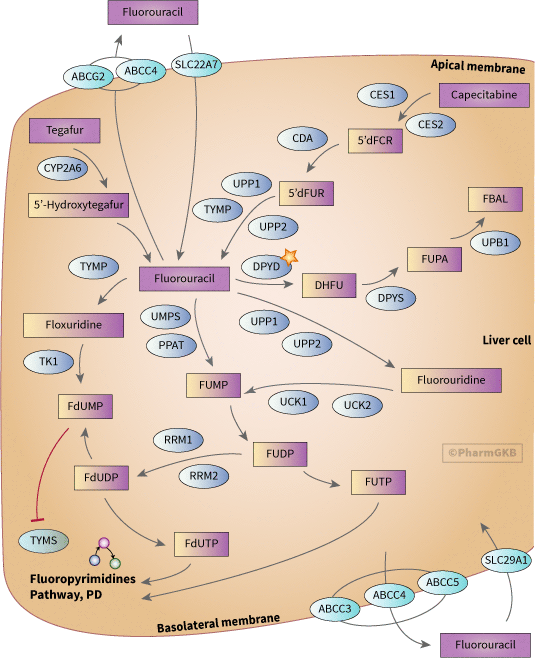
描述
Fluoropyrimidine 是一類廣泛用於治療癌症的抗代謝藥物,包括結腸直腸癌、乳腺癌及呼吸消化道癌症。此圖顯示了參與 藥物動力學 5-fluorouracil (5-FU)、capecitabine 和 tegafur 的候選基因。
5-FU 通常以靜脈注射的方式給予,超過 80% 的藥物在肝臟中被代謝 [Article:2656050]。capecitabine 是 5-FU 的口服前藥,能夠不經改變地通過腸壁,並在肝臟中轉化為 5'dFCR,然後由羧基酯酶和胞苷脫胺酶分別轉化為 5'-deoxy-5-fluorouridine (5'dFUR) [Articles:9849491, 18172246]。5'dFUR 隨後通過胸苷磷酸化酶或尿苷磷酸化酶轉化為 5-FU [Articles:9849491, 11956089]。tegafur 是另一種 5-FU 的前藥,經 CYP2A6 轉化為不穩定的中間體 5-hydroxytegafur,該中間體自發分解形成 5-FU [Article:18172246]。
5-FU 的代謝途徑有多種,其中一些會導致藥物的活化和藥效作用。5-FU 代謝的限速步驟是二氫嘧啶脫氫酶 (DPYD) 將 5-FU 轉化為 dihydrofluorouracil (DHFU) [Articles:14555507, 1272473]。DHFU 隨後由二氫嘧啶酶 (DPYS) 和 β-脲基丙酸酶 (UPB1) 分別轉化為 fluoro-beta-ureidopropionate (FUPA) 和 fluoro-beta-alanine (FBAL) [Article:14555507]。此 途徑 中酶的缺乏可能導致嚴重甚至致命的 5-FU 毒性。幾種 DPYD 的變異與毒性相關,包括(詳情請參見 DPYD VIP 和整理的註解)。DPYS 的變異也顯示出對 5-FU 毒性的影響。一種罕見變異 DPYS:833G>A (DPYS:Gly278Asp) 在第 5 外顯子中被證實是接受 5-FU 治療的荷蘭患者中嚴重毒性的決定性變異 [Article:14555507]。變異 DPYS:1635delC 和 DPYS:Leu7Val 在體外顯示出活性降低 [Article:18075467]。為了調節 Fluoropyrimidine 的活性,可以共同給予 DPYD 的抑制劑,如尿嘧啶和恩利尿嘧啶。這可以減緩 5-FU 的降解並改善反應率 [Article:12724731]。
5-FU 活化的主要機制是轉化為 fluorodeoxyuridine monophosphate (FdUMP),該物質抑制胸苷酸合成酶 (TYMS),這是葉酸-同型半胱氨酸循環及嘌呤和嘧啶合成的重要組成部分。5-FU 轉化為 FdUMP 可以通過胸苷酸磷酸化酶 (TYMP) 轉化為氟脫氧尿苷 (FUDR),然後由胸苷激酶轉化為 FdUMP,或間接通過 fluorouridine monophosphate (FUMP) 或氟脲苷 (FUR) 轉化為 fluorouridine diphosphate (FUDP),然後經由核糖核苷酸還原酶作用轉化為 FdUDP 和 FdUMP。FUDP 和 FdUDP 也可以轉化為 FUTP 和 FdUTP,並分別納入 RNA 和 DNA,這也有助於 Fluoropyrimidine 的藥效作用。
在使用 5-FU 和相關藥物時,一個重要的考量是腫瘤的藥物抗性發展。一些抗性機制涉及藥效基因候選者 (TYMS 和 P53) 的表達變化。藥物抗性也可能涉及藥物運輸的變化。關於 5-FU 的 藥物動力學 相關運輸蛋白的數據存在矛盾。在一項胰腺腫瘤的研究中,SLC29A1 的表達與生存率無關 [Article:18992248],但在另一項胰腺腫瘤細胞系的研究中,其表達與抗性/敏感性相關 [Article:17695509]。在一個體外表達系統中報導了 5-FU 的運輸 SLC22A7 [Article:15901346]。幾種運輸蛋白已被認為與 5-FU 抗性有關,包括 ABCG2 [Article:18820913][Article:18837291],ABCC3、ABCC4 和 ABCC5 [Article:19077464]。