CPIC 指南註解:fosphenytoin、phenytoin 與 CYP2C9、HLA-B 基因
摘要
Phenytoin/fosphenytoin 禁忌用於具有 HLA-B*15:02 變異的個體 等位基因("HLA-B*15:02-positive"),因為顯著增加了 phenytoin 誘發的皮膚不良反應的風險 史蒂芬-強森症候群及毒性表皮壞死溶解症(SJS/TEN)。此外,具有 CYP2C9 弱代謝型(Poor metabolizer) 表現型 或 CYP2C9 活性評分為 1 的患者可能需要減少 phenytoin/fosphenytoin 的劑量。
為特定註釋指定基因型或表現型
註釋
此註釋基於CPIC® 指南,針對 phenytoin 和 CYP2C9 及 HLA-B。
2020年8月
2020年8月線上提前發表
- 2020年CPIC指南對phenytoin的更新已在《Clinical Pharmacology and Therapeutics》期刊發表。審查了2014年4月至2019年8月間發表的文獻,並更新了建議和補充信息。這包括使用活性評分系統對CYP2C9 等位基因功能分配的更新。
- 此指南適用於:
- 成人患者
- 兒科患者
- 2020年phenytoin/fosphenytoin劑量指南更新摘錄:
- 「CYP2C9*2/*2雙倍型(AS=1)現在被翻譯為IM 表現型組(原本翻譯為PM)。此變更基於數據...顯示CYP2C9*1/*3(AS=1)和CYP2C9*2/*2對代謝比和劑量需求有相似的影響。此外,CYP2C9*3和具有相似臨床效果和功能的等位基因被歸類為‘功能缺失’等位基因,AS計算值為0。這基於CYP2C9*3/*3,代表代謝活性最低和藥代動力學清除最慢的雙倍型。其他具有相似低功能的等位基因也被歸類為‘功能缺失’。」
- 「大部分證據(總結於表S1)將HLA-B*15:02與phenytoin誘發的SJS/TEN聯繫起來,是在兒童和成人中產生的。因此,上述建議不論CYP2C9基因型、個人年齡、種族或祖源而制定。對於CYP2C9 IM或PM的兒科患者,建議根據表3和治療藥物監測(TDM)進行劑量調整。雖然對CYP2C9 等位基因對兒科患者群體中phenytoin代謝影響的數據有限,但沒有令人信服的數據表明CYP2C9多態性會在兒童中與成人不同地影響phenytoin代謝。因此,兒科建議是基於成人數據推斷的。新生兒和年長兒科患者的特殊考量,包括劑量變異性高和劑型,以及來自兒科群體的證據,已包含在補充資料中(詳情見補充資料的兒科部分)。」
- 下載並閱讀:
表1:基於HLA-B*15:02和CYP2C9的phenytoin/fosphenytoin建議
改編自2020年指南更新的表1、2和3。
- HLA-B*15:02「陽性」:1或2個HLA-B*15:02 等位基因
- HLA-B*15:02「陰性」:未報告HLA-B*15:02 等位基因
| HLA-B*15:02基因型 | CYP2C9 表現型 | CYP2C9活性評分 | CYP2C9基因型 | 影響 | 治療建議 | 建議分類 | 考量 |
|---|---|---|---|---|---|---|---|
| HLA-B*15:02「陽性」 | 任何CYP2C9 表現型 | 任何CYP2C9活性評分 | 任何CYP2C9基因型 | phenytoin誘發的SJS/TEN風險增加 | 如果患者是phenytoin初次使用者,請勿使用phenytoin/fosphenytoin。避免使用carbamazepine和oxcarbazepine。 可選建議建議:如果患者之前已連續使用phenytoin超過三個月且未發生皮膚不良反應,未來可謹慎考慮使用phenytoin。藥物誘發SJS/TEN的潛伏期在連續給藥和遵從治療的情況下較短(4-28天),通常在給藥三個月內發生。 |
強烈建議 | 其他芳香族抗癲癇藥,包括eslicarbazepine、lamotrigine和phenobarbital,與HLA-B*15:02 等位基因的SJS/TEN聯繫證據較弱;然而,選擇替代藥物時仍應謹慎。先前對phenytoin的耐受性並不表示對其他芳香族抗癲癇藥的耐受性。 |
| HLA-B*15:02「陰性」 | CYP2C9 正常代謝型(Normal metabolizer) | 2 | 攜帶兩個功能正常 等位基因的個體;示例雙倍型:*1/*1 | 正常phenytoin代謝 | 不需要從典型劑量策略中調整。後續劑量應根據治療藥物監測(TDM)、反應和副作用進行調整。HLA-B*15:02陰性測試並不排除phenytoin誘發的SJS/TEN風險,應根據標準實踐仔細監測患者。 | 強烈建議 | |
| HLA-B*15:02「陰性」 | CYP2C9 中間代謝型(Intermediate metabolizer) (AS 1.5) | 1.5 | 攜帶一個功能正常 等位基因加上一個功能減弱 等位基因的個體;示例雙倍型:*1/*2 | phenytoin代謝略有減少;然而,這似乎不會轉化為增加的副作用。 | 不需要從典型劑量策略中調整。後續劑量應根據治療藥物監測(TDM)、反應和副作用進行調整。HLA-B*15:02陰性測試並不排除phenytoin誘發的SJS/TEN風險,應根據標準實踐仔細監測患者。 | 中等建議 | |
| HLA-B*15:02「陰性」 | CYP2C9 中間代謝型(Intermediate metabolizer) (AS 1) | 1 | 攜帶一個功能正常 等位基因加上一個功能減弱 等位基因或兩個功能減弱 等位基因的個體;示例雙倍型:*1/*3, *2/*2 | phenytoin代謝減少,較高的血漿濃度將增加毒性的可能性。 | 首次劑量使用典型初始或速效劑量。後續劑量使用比典型維持劑量少約25%。後續劑量應根據治療藥物監測(TDM)、反應和副作用進行調整。HLA-B*15:02陰性測試並不排除phenytoin誘發的SJS/TEN風險,應根據標準實踐仔細監測患者。 | 中等建議 | |
| HLA-B*15:02「陰性」 | CYP2C9 弱代謝型(Poor metabolizer) | 0-0.5 | 攜帶一個功能缺失 等位基因加上一個功能減弱 等位基因或兩個功能缺失 等位基因的個體;示例雙倍型:*2/*3, *3/*3 | phenytoin代謝減少,較高的血漿濃度將增加毒性的可能性。 | 首次劑量使用典型初始或速效劑量。後續劑量使用比典型維持劑量少約50%。後續劑量應根據治療藥物監測(TDM)、反應和副作用進行調整。HLA-B*15:02陰性測試並不排除phenytoin誘發的SJS/TEN風險,應根據標準實踐仔細監測患者。 | 強烈建議 | |
| HLA-B*15:02「陰性」 | 無法判定 | N/A | 攜帶一個或兩個CYP2C9 功能未知或功能尚未確定 等位基因的個體。 | N/A | 無建議 | N/A |
2014年11月
2014年8月線上接受文章預覽;2014年9月線上提前發表
- 關於phenytoin劑量中使用藥物基因組學測試的指南已由臨床藥物基因體學實施聯盟(CPIC)發表於《Clinical Pharmacology and Therapeutics》期刊。
- 2014年phenytoin劑量指南摘錄:
- 「對於 CYP2C9 中間代謝型(intermediate metabolizers),可考慮將建議的起始維持劑量至少降低 25%,後續維持劑量則應依治療藥物監測(therapeutic drug monitoring, TDM)結果及臨床反應進行調整。 對於 CYP2C9 弱代謝型(poor metabolizers),則建議考慮將起始維持劑量至少降低 50%,後續維持劑量同樣應依治療藥物監測或臨床反應進行調整。」
- 「不論 CYP2C9 基因型,以及個體的族群背景或年齡,只要 HLA-B15:02 檢測結果為陽性,即建議考慮使用 carbamazepine 與 phenytoin 以外的抗癲癇藥物,除非治療原發疾病的潛在效益明顯高於相關風險。⋯⋯替代藥物如 oxcarbazepine、eslicarbazepine acetate 及 lamotrigine,亦有部分證據顯示其與 HLA-B15:02 等位基因相關的 SJS/TEN 風險,因此在選擇 phenytoin 的替代用藥時,應審慎評估並特別留意相關風險。」
- 下載並閱讀:
表2:基於HLA-B和CYP2C9 表現型/基因型的phenytoin/fosphenytoin治療建議
改編自2014年指南手稿的表1和2。
- HLA-B*15:02「陽性」:1或2個*15:02 等位基因s
- HLA-B*15:02「陰性」:未報告HLA-B*15:02 等位基因
| CYP2C9 表現型 | CYP2C9基因型 | HLA-B*15:02「陽性」- 影響 | HLA-B*15:02「陽性」- 治療建議 | HLA-B*15:02「陽性」- 建議分類 | HLA-B*15:02「陰性」- 影響 | HLA-B*15:02「陰性」- 治療建議 | HLA-B*15:02「陰性」- 建議分類 |
|---|---|---|---|---|---|---|---|
| CYP2C9 廣泛代謝型(Extensive metabolizer) | 正常活性~91%的患者;攜帶2個正常活性等位基因的個體;示例雙倍型:*1/*1 | phenytoin誘發的SJS/TEN風險增加 | 如果患者是phenytoin初次使用者b,請勿使用phenytoin/fosphenytoin。 | 強烈建議 | 正常phenytoin代謝 | 以推薦的維持劑量開始治療d。 | 強烈建議 |
| CYP2C9 中間代謝型(Intermediate metabolizer) | 異型合子~8%的患者;攜帶一個正常活性等位基因加上一個功能減弱 等位基因的個體;示例雙倍型:*1/*3, *1/*2 | phenytoin誘發的SJS/TEN風險增加 | 如果患者是phenytoin初次使用者b,請勿使用phenytoin/fosphenytoin。 | 強烈建議 | phenytoin代謝減少,較高的血漿濃度將增加毒性的可能性。 | 考慮將推薦的起始維持劑量減少25%d。後續劑量應根據治療藥物監測(TDM)和反應進行調整。 | 中等建議 |
| CYP2C9 弱代謝型(Poor metabolizer) | 同型合子(Homozygous)變異~1%的患者;攜帶2個功能減弱 等位基因的個體;示例雙倍型:*2/*2, *3/*3, *2/*3 | phenytoin誘發的SJS/TEN風險增加。 | 如果患者是phenytoin初次使用者b,請勿使用phenytoin/fosphenytoin。 | 強烈建議 | phenytoin代謝減少,較高的血漿濃度將增加毒性的可能性。 | 考慮將推薦的起始維持劑量減少50%d。後續維持劑量應根據治療藥物監測(TDM)和反應進行調整。 | 強烈建議 |
b 如果患者之前已使用phenytoin超過3個月且未發生皮膚不良反應,請謹慎重新使用phenytoin。根據CYP2C9基因型(如果已知)調整劑量。 d 根據患者的臨床特徵推薦維持劑量。
Acetaminophen 途徑 (治療劑量), 藥物動力學
概括
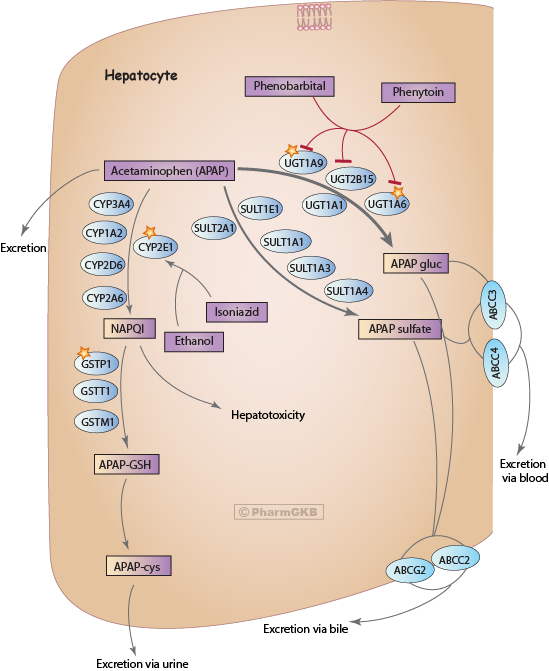
肝臟中 acetaminophen 代謝與運輸之示意圖
描述
acetaminophen(N-乙醯對氨基酚,APAP或PARAcetamol,PARA)因其鎮痛和退燒特性而廣泛應用於許多成人和兒童的非處方配方中[Articles:21054454,23719833]。APAP可通過前藥pheNACetin的O-去烷基化在體內合成,該藥物因腎毒性和致癌性而被撤回市場[Article:7002186]。在治療性成人劑量1-2 g/天下,口服APAP適用於發燒及輕度至中等建議急性疼痛狀況[Article:23719833。通過靜脈途徑給予[[KEEP_fc662ABC]]的使用越來越普遍,並已被用作安全有效的退燒和鎮痛劑[Article:25521845]。成人的最大建議治療劑量為4 g/天,兒童為50-75 mg/kg/天。成人單次劑量超過7 g及兒童超過150 mg/kg被認為對肝臟和腎臟具有潛在毒性,因為其活性代謝物N-乙醯對苯醌亞胺(NAPQI)[Article:22998987]。
代謝
肝臟,以及腎臟和腸道(程度較小),是acetaminophen代謝的主要器官[Article:11215692]。在經過治療劑量後,APAP主要轉化為藥理活性NAC的葡萄糖醛酸酯(APAP-gluc,52-57%尿液代謝物)和硫酸酯(APAP sulfate,30-44%)的結合物,少量被氧化為反應性代謝物NAPQI(5-10%)。少於5%的APAP以未改變的形式排泄[Article:6343056]。NAPQI具有高度反應性,主要負責acetaminophen引起的肝毒性。NAPQI的解毒過程是通過其與谷胱甘肽的巰基(GSH)結合形成APAP-GSH,最終以半胱氨酸和巰基酸結合物(APAP-cys)排泄於尿液中[Articles:22998987,11215692]。acetaminophen的分佈涉及肝臟、腎臟和腸道之間的代謝物複雜的跨器官運輸,通過膽汁和血液最終排泄於尿液和糞便中[Article:11215692]。大多數葡萄糖醛酸酯和硫酸酯代謝物從肝臟通過血液運輸到腎臟,而部分APAP-gluc出現在膽汁中,隨後運輸到腸道。腎臟是APAP硫酸酯的主要分佈部位,無論是通過直接排泄還是通過進一步的生物轉化後再經腎臟排泄。雖然大多數NAPQI是在肝臟中形成,但腎臟也會將APAP代謝為毒性代謝物,並將APAP的半胱氨酸結合物釋放到膽汁和血液中以進一步排泄於尿液中[Article:11215692]。
在超過治療劑量的APAP劑量(超過4 g/天)時,硫酸化途徑變得飽和,而葡萄糖醛酸化和氧化增加,且少量以未改變的形式排泄。在高毒性劑量的APAP後,葡萄糖醛酸化也會飽和,且更高比例的藥物以未改變的形式排泄(約10%),並被氧化為NAPQI(>15%)(/途徑/PA166117881)。過量的NAPQI最終會耗竭GSH的儲存,並開始通過與細胞蛋白上的半胱氨酸基團結合形成蛋白質加合物。NAPQI主要針對線粒體蛋白和離子通道,導致能量產生的喪失、離子失衡和細胞死亡[Articles:22998987,11215692,14625346]。根據動物研究,N-乙醯半胱氨酸(NAC)被證明是acetaminophen過量的有效解毒劑[Article:7469630]。NAC補充GSH的儲存,清除線粒體中的活性氧物質,並增強硫酸化代謝途徑(/途徑/PA166117881)。如果在急性過量後8-10小時內給予,NAC可將肝毒性風險降低至5%以下。總體而言,NAC可防止肝損傷、腎衰竭和死亡,並是APAP中毒的首選治療[Articles:22998987,11215692,6343056,3059186]。極高劑量的APAP會導致嚴重肝損傷,伴隨著葡萄糖醛酸化和硫酸化能力的顯著降低[Article:6343056]。在致命的中央小葉肝壞死患者中,血漿和尿液中的葡萄糖醛酸代謝物幾乎無法檢測[Article:4788034]。
葡萄糖醛酸化途徑的acetaminophen代謝由UDP-葡萄糖醛酸轉移酶(UGT)催化。UGT使APAP分子更具水溶性,通過將葡萄糖醛酸基團從UDP-葡萄糖醛酸轉移[Articles:22998987,23462933]。在人類肝臟微粒體和培養的肝細胞中研究表明,UGT1A1、UGT1A6、UGT1A9和UGT2B15參與APAP的葡萄糖醛酸化[Articles:8494539,11714888,16696573,15933229]。UGT1A6在低APAP濃度下非常重要[Article:11714888],而UGT1A9和UGT1A1在毒性劑量下貢獻最大,UGT1A9在廣泛的藥理相關APAP濃度範圍內催化[Articles:11714888,16696573]。
一組細胞質酶,稱為硫酸轉移酶(SULT),負責acetaminophen的硫酸化。SULT將硫酸基團從底物PAPS轉移到APAP,使其更極性並易於排泄[Article:23462933]。使用人類血小板匀浆作為肝臟外源性代謝的模型,首次顯示SULT1A1和SULT1A3/4催化APAP的硫酸化[Article:6950087]。人類SULT1A3和SULT1A4基因非常相近,編碼相同的SULT蛋白[Article:14676822]。除了SULT1A1和1A3/4外,人類胎肝中的APAP的硫酸化還由SULT1E1和SULT2A1進行[Article:18232020]。這項研究顯示,在胎肝中,SULT1A3/4在APAP的硫酸化中起主要作用;然而,在出生後的發展中,APAP主要由SULT1A1和SULT2A1硫酸化,而SULT1A3/4的活性減少[Article:18232020]。
細胞色素P450酶催化acetaminophen的氧化,生成反應性代謝物NAPQI [Articles:23462933,11215692]。特定CYP亞型對APAP的生物活化的具體貢獻因藥物濃度而異。在人類肝臟微粒體中,首次報導CYP2E1和CYP1A2將高劑量的APAP轉化為NAPQI [Article:2729995]。後續研究使用純化的人類蛋白或人類肝臟微粒體和特定抑制劑確認CYP2E1在毒性劑量的APAP生物活化中的作用,但也報導了CYP2A6的參與[Articles:9548799,11866476]。對健康人類志願者進行的研究,使用CYP2E1抑制劑二硫仿進一步確認CYP2E1在APAP氧化中的作用[Article:10741631]。使用人類肝臟微粒體和人類重組CYP2D6,報導該酶僅在非常高的毒性劑量下氧化APAP,當血漿APAP濃度達到2 mM時[Articles:11095574,19219744]。CYP3A4在APAP代謝中的作用存在爭議,研究結果從無顯著貢獻到在APAP氧化中的主要作用不等[Articles:10741631,19219744,8387297,8374050]。
NAPQI與GSH的結合通過自發過程和由谷胱甘肽-S-轉移酶(GST)催化的酶促反應進行[Article:3395122]。非酶促反應產生GSH結合物,3-(谷胱甘肽-S-基)-acetaminophen(APAP-GSH);還有還原產物,自由的APAP;以及氧化產物,谷胱甘肽二硫化物(GSSG)。GST反應產生APAP-GSH和自由的APAP。人類細胞質GST家族由七個不同類別的酶組成,每個類別內有多種遺傳變異[Article:23201197]。人類體外研究顯示,與分離的肝臟和胎盤GST進行的研究表明,GSTP1是NAPQI與GSH結合的最有效催化劑,其次是GSTT1和GSTM1 [Article:3395122]。在NAPQI的還原反應中,最有效的人類轉移酶是GSTT1,其次是GSTM1和GSTP1。
運輸
acetaminophen的分佈和排泄取決於其通過不同細胞類型的運輸。與母藥不同,acetaminophen代謝物的移動需要運輸蛋白。acetaminophen與兩個超家族的運輸蛋白的共同藥物載體的相互作用已在溶質載體運輸蛋白(SLC)和ATP-結合盒(ABC)運輸蛋白的背景下進行了探討[Articles:17627974,22406294,21389119,18789319]。ABC運輸蛋白介導底物從細胞的外排,而SLC運輸蛋白則負責底物進入細胞[Articles:22013971,16586096]。APAP-gluc和硫酸酯的排泄進入膽汁涉及位於肝細胞管腔膜的ABCC2和ABCG2載體。APAP-gluc進入血液的移動依賴於ABCC3運輸蛋白,而硫酸代謝物則依賴於位於肝細胞的窦側的ABCC3和ABCC4。此外,ABCB1、ABCC1和ABCC5運輸蛋白可能參與acetaminophen的排泄,這在毒性acetaminophen攝入後表現出其表達的變化[Article:17627974]。來自acetaminophen過量的患者的肝臟顯示ABCC1和ABCC4 mRNA的上調,以及ABCB1、ABCG2、ABCC4和ABCC5的蛋白質水平升高。排出運輸蛋白的表達增加可能是為了防止細胞中毒性代謝物的積累,並防止進一步的肝損傷。與此假設一致的是肝細胞增殖的增加以及上調的運輸蛋白與快速增殖的肝細胞區域的共定位[Article:17627974]。這些對acetaminophen的毒性水平的適應性反應導致了對肝臟的重複SULT的獲得性抵抗。這一現象類似於在實驗動物中觀察到的自我保護,初次接觸acetaminophen的亞毒性劑量可保護啮齒動物免受隨後致死劑量的影響[Articles:473230,24973094]。個別病例報告表明,人類受試者在未造成肝損傷的情況下,對重複和高劑量的acetaminophen可能會發展出耐受性[Articles:9918922,7748624]。雖然這些患者對acetaminophen過量的肝毒性抵抗的機制尚未完全闡明,但通過上調外排運輸蛋白的自我保護可能是導致對該藥物的慢性和致死劑量耐受性的原因。
SLC運輸蛋白由兩個基因超家族組成,SLC22A超家族包含有機陽離子運輸蛋白(OCTs)和有機陰離子運輸蛋白(OATs),而SLCO超家族則包括有機陰離子運輸多肽(OATP)。OATP主要運輸大型、疏水性的有機陰離子,而OATs則運輸小型和親水性分子;OCTs則介導陽離子的運動[Article:22013971]。使用穩定表達人類運輸蛋白的細胞系,評估acetaminophen與hOATs和hOCTs的相互作用[Article:12388633]。acetaminophen抑制由hOAT1(SLC22A6)、2(SLC22A7)、3(SLC22A8)和4(SLC22A9)介導的有機陰離子攝取。OCT1(SLC22A1)和2(SLC22A2)未介導acetaminophen的攝取,但可以被其抑制,這表明acetaminophen可能會干擾依賴這些運輸蛋白的其他藥物的排除[Article:12388633]。關於OATP家族,體外測試顯示acetaminophen未與OATP1B1(SLCO1B1)或OATP1B3(SLCO1B3)運輸蛋白相互作用[Article:21389119]。
藥物交互作用
許多藥物已被報導與acetaminophen相互作用,導致其毒性加劇[Articles:15933229,3511825,8470644,2049229,17151811,3346037]。幾個病例報告建議,長期使用抗癲癇藥的癲癇患者表現出增加的acetaminophen引起的肝毒性[Articles:3346037,760753,1354974,6465717]。在大多數情況下,phenytoin或phenobarbital的慢性使用在acetaminophen過量後增強了毒性的臨床特徵[Articles:3346037,760753,1354974]。然而,若與phenobarbital同時攝取,即使是低治療劑量劑量的acetaminophen也會加劇其毒性[Article:6465717]。有研究表明,癲癇患者因藥物的首過代謝增加而表現出較低的acetaminophen生物利用度[Article:760753]。使用人類肝細胞作為急性藥物-藥物相互作用的模型,報導phenytoin和phenobarbital抑制acetaminophen的葡萄糖醛酸化[Articles:16696573,15933229]。每種藥物單獨或聯合直接阻止UGT1A6、UGT1A9和UGT2B15,當與acetaminophen共同培養時[Articles:16696573,15933229]。這表明當葡萄糖醛酸化受到損害時,acetaminophen會轉向其他途徑,導致反應性代謝物NAPQI的過量產生。可以合理推測,這一現象是導致癲癇患者acetaminophen引起的毒性加劇的原因。
許多藥物,包括ethanol和isoniazid,在其代謝過程中誘導CYP450的同工酶[Articles:8354023,184555。抗結核藥物isoniazid誘導CYP2E1,這對[[KEEP_c61c3ABC]]的氧化代謝至關重要途徑。與isoniazid共同給藥的報導顯示,增加了acetaminophen的氧化,促進了GSH的耗竭和NAPQI的形成,最終導致肝毒性的增加[Articles:8470644,2049229,2240884]。CYP2E1也被ethanol顯著上調,且在酗酒者中肝毒性已被充分記錄[Article:3511825]。低至中等建議劑量的acetaminophen與大量飲酒結合會導致肝酶異常、黃疸和凝血功能障礙。綜合來看,接受isoniazid治療或過量飲酒的受試者在使用acetaminophen時應謹慎,以避免因CYP2E1誘導而導致的肝毒性。
phenytoin 途徑, 藥物動力學
概括
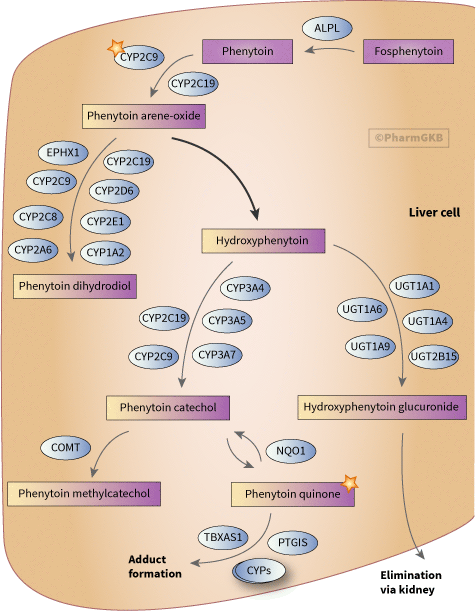
人類肝細胞中參與 fosphenytoin 和 phenytoin 代謝的基因示意圖。
描述
背景
phenytoin 是在 USA 中最常被處方的抗癲癇藥物之一(約 52% 的 AED 處方,相較於 19% 的丙戊酸、11% 的卡馬西平和 7% 的苯巴比妥)[Article:19855097。它具有狹窄的治療範圍和廣泛的個體間清除率變異,因此,治療藥物監測(TDM) 對於 TEN 是必要的。phenytoin 的不良反應範圍從輕微(例如牙齦增生)到嚴重和危及生命(例如史蒂文斯-約翰遜綜合症、SJS 和毒性表皮壞死症 TEN)以及致畸(例如出生缺陷)。fosphenytoin 是 phenytoin 的水溶性前藥。CPIC 指南] 禁止在具有 HLA-B*15:02 變異的個體中使用 fosphenytoin/phenytoin(“HLA-B*15:02 陽性”),因為這會顯著增加 phenytoin 引起的皮膚不良反應的風險 史蒂芬-強森症候群及毒性表皮壞死溶解症(SJS/TEN)。此外,具有 CYP2C9 弱代謝型(Poor metabolizer) 表現型 或 CYP2C9 活性得分為 1 的患者可能需要減少 phenytoin/fosphenytoin 的劑量。
藥物動力學
代謝酶
phenytoin 主要代謝為無活性的 hydroxyphenytoin,5-(4'-羥基苯基)-5-苯基氨基脲或 p-HPPH [Article:9798756]。高達 90% 的 phenytoin 被代謝為 p-HPPH,然後經葡萄糖醛酸化並排泄到尿液中 17576806。形成了兩種立體異構體的 p-HPPH: (R)-p-HPPH 和 (S)-p-HPPH,且相對量存在相當大的個體間變異 [Articles:16815679, 3572817]。當反應由 CYP2C19 催化時,(R)-p-HPPH 和 (S)-p-HPPH 的比率約為 1:1 [Article:8971149]。然而,當反應由 CYP2C9 催化時,S 異構體的形成比率可高達 40:1 [Articles:16815679, 8971149]。因此,立體異構體的相對比率可用於 表現型 CYP2C9 和 CYP2C19 的基因組變異。
p-HPPH 的形成被認為是通過反應性芳香烴氧化物中間體進行的 [Article:6130920],而“芳香烴氧化物假說”是大多數報告 phenytoin 過敏反應、史蒂文斯-約翰遜綜合症/毒性表皮壞死症 (SJS/TEN)、肝毒性和其他形式的特異性毒性的主要機制。芳香烴氧化物也可以通過環氧化物水解酶轉化為 phenytoin dihydrodiol [Article:9798756]。在體外實驗中,二氫二醇的形成也被證明是由 CYP1A2、CYP2C19、CYP2E1、CYP2A6、CYP2D6、CYP2C8、CYP2C9 和 CYP3A4 催化的 [Article:11038165]。二氫二醇代謝物也可以轉化為兒茶酚 [Article:9798756]。
hydroxyphenytoin 可以通過幾種 P450 酶轉化為兒茶酚,3'-4'-二HPPH。CYP2C19 被發現是兒茶酚形成的最有效催化劑,但 CYP2C9 和 CYP3A4 可能由於其在肝臟中的相對優勢而負責大部分轉化 [Articles:11038165, 10901705]。 CYP3A5、CYP3A7、CYP2D6 和 CYP2B6 也被證明在體外催化兒茶酚的形成 [Articles:11038165, 10901705]。儘管在肝臟中的表達有限,CYP2C18 被報導在皮膚中表達,並已被證明催化主要和次要的羥基化步驟 [Article:16359177],這可能與皮膚不良藥物反應(ADRs)有關。
兒茶酚自發氧化形成反應性醌代謝物,該代謝物可以通過 NQO1 代謝回兒茶酚。兒茶酚也可以通過 COMT 代謝為甲基兒茶酚,然後排泄到尿液中 [Article:9798756]。
hydroxyphenytoin 由 UGTs 葡萄糖醛酸化,特別是 UGT1A1、UGT1A4、UGT1A6 和 UGT1A9。有提議認為這種葡萄糖醛酸化可以防止 hydroxyphenytoin 被過氧化物酶轉化為一種有毒的反應性代謝物,該代謝物可以氧化蛋白質、脂質和 DNA [Article:15855726]。p-HPPH 的葡萄糖醛酸化是立體特異性的 擇期,UGT1A1 優先對 S 異構體進行葡萄糖醛酸化,而 UGT1A9 和 UGT2B15 則作用於 R 異構體 [Article:17576806]。
對 phenytoin 的 ADRs 通常分為兩類:劑量(或濃度)依賴性毒性和特異性過敏反應。如下面更詳細討論的,濃度依賴性 ADRs 與 para-羥基化受損和 phenytoin 的過度積累有關。對於哪些藥物及其代謝物最常涉及產生過敏反應仍存在一些爭議。phenytoin 和 p-HPPH 已被證明與多種內源性酶形成加合物 [Article:16359177]。在過敏患者的血清中觀察到識別 CYP3A4、PTGIS 和 TBXAS1 的抗體 [Article:9798756]。
運輸
對於 [[KEEP_eaBBB4b9]],最廣泛討論的運輸蛋白是 ABCB1。ABCB1 已在體外實驗中顯示能夠在細胞系中跨梯度運輸 phenytoin [Articles:20417647, 18824002, 18408562]。ABCB1 在血腦屏障(BBB)的作用被認為是對 AEDs(包括 phenytoin)的抗藥性機制。各種研究探討了 ABCB1 變異對抗藥性的影響(見 藥物基因體學 部分)。ABCB1 在癲癇大腦中被證明過度表達 [Article:17576806]。COX-2 抑制劑已被證明能減少癲癇相關的 ABCB1 上調,並改善 phenytoin 的腦部運輸,防止動物模型中的抗藥性 [Article:19786037]。
在大鼠的實驗中,ABCC2 被認為在跨越 BBB 運輸 phenytoin 中起作用 [Article:12663688],但體外細胞系研究並未支持 phenytoin 通過 ABCC1、ABCC2 或 ABCC5 的運輸 [Article:20080116]。
藥效學
phenytoin 針對大腦中的電壓門控鈉通道 [Articles:10514834, 20643904]。電壓門控鈉通道是由一個大型糖基化的 α 次單元(約 260 kD)和兩個較小的 β 次單元(33-39 kD)組成的異源複合體。電壓門控鈉通道由 SCN 基因家族編碼,該家族的成員在心臟和骨骼肌以及周邊和中樞神經系統中表達。基因 SCN1A、SCN2A 和 SCN3A 編碼在大腦中表達的 α 次單元 [Article:17101538]。SCN1A 的突變與癲癇相關;已報導超過 500 種變異,包括與德拉維特綜合症(又稱嬰兒重度肌陣攣癲癇)相關的變異 (SMEI)[Article:20351042]。癲癇相關的變異也已在 SCN2A 中報導,但在 SCN3A 中記錄的變異較少 [Article:20351042]。
phenytoin 偏好性地在開放狀態下結合 SCN2A 通道 [Article:20643904]。據認為 phenytoin 在慢速放電率下對鈉通道的阻斷效果較差,允許正常的腦部活動,但抑制癲癇特徵的高頻重複放電 [Articles:20643904, 9212254]。因此,鈉通道中的變異可能會影響 phenytoin 的療效(見下文)。
藥物基因體學
幾項研究報導了基因組變異與劑量、代謝比率或血漿藥物水平之間的關聯;相對較少的研究探討了它們在藥物抗藥性和 ADRs 中的作用。大多數研究考察了 CYP2C9 的作用,少數研究考察了其他代謝酶、運輸蛋白和藥效學候選基因。
代謝酶變異
受損的 phenytoin para-羥基化首次於 1964 年報導 [Article:14161752],其特徵為 phenytoin 濃度增加、消除半衰期延長和中毒跡象 - 眼震、共濟失調和意識障礙 [Article:7351122][Article:6879646][Article:3180640]。
CYP2C9*3 (rs1057910 A>C) 與 phenytoin 的代謝減少相關,這在癲癇患者的藥物動力學研究中得到了體內和體外的證實 [Articles:16815679, 10901705, 21068649]。 CYP2C9*3 也與癲癇患者的劑量增加相關 [Article:15805193。對 CYP2C9*2 (rs1799853] C>T) 變異的研究結果相互矛盾。CYP2C9*2 在北美的一項研究中與癲癇患者的代謝減少相關 [Article:16815679],但在一項針對白人癲癇患者的研究中未與代謝或 phenytoin 劑量相關 [Articles:10901705, 15805193]。這一差異可能是由於與 CYP2C19*2 連鎖的 CYP2C9 啟動子中的其他變異所致 [Article:19855097]。A 等位基因 的 rs12782374G>A 和 rs71486745T>del 的缺失與癲癇患者的 phenytoin 劑量減少相關 [Article:19855097]。其他 CYP2C9 變異在黑人群體中存在,CYP2C9*5、*6、*8 和 *11 但不 CYP2C9*9,與 phenytoin 的代謝減少相關 [Article:16220110]。
最近一項針對印度亞洲人的研究顯示,phenytoin 在 CYP2C9*3 攜帶者的血漿中增加,並顯示出與濃度依賴性毒性風險增加相關,與 *1 同型合子相比 21068649。該研究還顯示 phenytoin 在 CYP2C9*2 攜帶者的血漿中增加,但雜合子並未顯示出顯著增加的藥物毒性風險。然而,該研究中唯一的 同型合子(Homozygous) CYP2C9*2 個體確實顯示出增加的藥物毒性風險 [Article:21068649]。該研究還指出,在營養不良的個體中 phenytoin 的代謝受損,CYP2C9 變異的影響更為明顯。一項小型研究(n=14)針對美國的 phenytoin 毒性案例(所有案例均為白人)顯示,CYP2C*1*3 和 CYP2C9*2*2 基因型的風險增加,儘管這些並不顯著 [Article:19617466]。
少數研究探討了 藥物基因體學 以外的基因對藥物的影響。針對 CYP2C19 rs4244285(存在於 *2 等位基因)的癲癇患者的研究顯示,與 *1 同型合子相比,雜合子代謝 phenytoin 減少 [Article:16815679。目前尚未觀察到 CYP2C19 變異對藥物療效或毒性的影響,儘管有幾項研究報導了其他藥物動力學候選基因對臨床結果的影響。CYP1A1 rs2606345] A 等位基因 與癲癇女性在使用卡馬西平、苯巴比妥、phenytoin 或丙戊酸治療時癲癇發作風險增加相關,而與 C 等位基因 [Article:21121773。然而,這一關聯在男性中未見。兩個母體 SNP 在 EPHX1 中(rs2234922 G 和 rs1051740] T)與女性在懷孕第一三個月接觸 phenytoin 時顱面畸形的風險相關 [Article:19952982]。
運輸蛋白變異
一些證據表明,著名的 ABCB1 變異 3435C>T rs1045642 影響血漿藥物水平和藥物抗藥性。單倍型 三個 ABCB1 變異的組合,包括 rs1045642,改變了 phenytoin 抑制的標記底物的運輸 [Article:18408562。在另一項研究中,包含 rs1045642] T 等位基因 的不同 單倍型 與健康的黑人非洲志願者的血漿藥物水平增加相關 [Article:16220110。兩者 單倍型 也包含 1236C>T (rs1128503)。在一項針對英國癲癇患者的研究中,雖然具體的 AED 並未定義,但 rs1045642] CC 基因型與藥物抗藥性相關 [Article:12686700]。此外,一項針對埃及癲癇患者的研究顯示,C 等位基因 攜帶者對 phenytoin 的抗藥性可能性增加 17529887。進一步的研究,包括一項元分析 [Article:19178561,未能重複與 rs1045642] 的關聯,儘管這些研究中的許多患者使用的是各種 AED 而非單一的 phenytoin。元分析中包含的研究也在白人和亞洲群體的子組中進行了分析,結果整體仍為陰性,但未考慮黑人或非裔美國人的研究,這可能會影響不同的 單倍型。
藥效學變異
對於 phenytoin 的 藥效學 候選基因進行了一些研究。SCN1A rs3812718 的 T 等位基因 與癲癇患者的 phenytoin 劑量增加相關,相較於 等位基因 C [Articles:15805193, 17001291。體外研究支持這一關聯,顯示 rs3812718] 影響 SCN1A 編碼的 NaV1.1 的可變剪接。C 等位基因 與 Na(V) 1.1-5N 剪接變異體的表達增加相關,而 TT 基因型幾乎不表達 Na(V) 1.1-5N,僅表達 Na(V) 1.1-5A [Article:17436242]。NaV1.1-5N 剪接變異體通道與 phenytoin 的敏感性增加相關,相較於 NaV1.1-5A 剪接變異體通道 [Article:21453355]。
SCN2A SNP rs2304016 A 等位基因 與中國癲癇患者對 AEDs(包括 phenytoin)的藥物抗藥性相關 [Article:18784617]。
主要組織相容性位點變異
雖然主要針對另一種 AED 卡馬西平進行了更深入的研究,但主要組織相容性位點變異也被調查其在 phenytoin 引起的 ADRs 中的作用。最受研究的變異是高度複雜的 單倍型,跨越 HLA-B 基因及其周圍區域,對應於血清型 表現型,並可能在不同人群中由不同的 SNP 標記。HLA-B*15:02 等位基因 與幾個亞洲人群中對卡馬西平的嚴重 ADRs 相關 ,詳情見卡馬西平 途徑。
HLA-B*15:02 等位基因 與泰國人群中 phenytoin 引起的 SJS 相關 [Article:18637831],並且也與中國亞洲人群中 phenytoin 引起的 SJS 和 TEN 相關 [Article:20235791]。其他 MHC 位點 等位基因 HLA-B*13:01、Cw*08:01 和 DRB1*16:02 也顯示與 phenytoin 引起的 SJS/TEN 在漢族中國人中相關 [Article:20235791]。
結論
藥物動力學 的 phenytoin 研究相當充分,基因組變異對血漿藥物水平和代謝的影響已在健康人群和癲癇患者中進行了研究。然而,較少的研究顯示這些代謝變化如何影響藥物反應、抗藥性和 ADRs。需要進一步研究考慮多個變異和 單倍型 及其在明確定義的人群中的影響,最好是針對 phenytoin 單獨進行。