CPIC 指南註解:fluvoxamine 與 CYP2D6 基因
摘要
CPIC 劑量指引對於 選擇性血清素再回收抑制劑(SSRI) fluvoxamine 建議,對於 CYP2D6 弱代謝型(Poor metabolizer),考慮將建議的起始劑量減少 25-50%,並採用較慢的劑量調整計畫,或使用不經 CYP2D6 代謝的替代藥物。
為特定註釋指定基因型或表現型
註釋
此註釋基於CPIC®指引,針對CYP2D6、CYP2C19、CYP2B6、SLC6A4和HTR2A基因型與血清素再攝取抑制劑抗抑鬱藥。這是對先前CPIC®指引的更新,涉及選擇性血清素再回收抑制劑(SSRI)和CYP2D6及CYP2C19。
2023年2月
-
2015年CPIC®指引針對選擇性血清素再回收抑制劑(SSRI)和CYP2D6及CYP2C19進行了更新,納入了更多基因和藥物。現在被稱為CPIC®指引,針對CYP2D6、CYP2C19、CYP2B6、SLC6A4和HTR2A基因型與血清素再攝取抑制劑抗抑鬱藥。2015年1月至2022年6月間發表的有關fluvoxamine和CYP2D6的文獻已被審查,並更新了建議和補充信息。
-
指引摘錄:
- 「目前數據不足以評估CYP2D6超快速代謝對fluvoxamine暴露或臨床結果的影響。因此,對於CYP2D6超快速代謝者,未提供venlafaxine或fluvoxamine的劑量建議。」
- 「在給予相似劑量時,CYP2D6慢代謝者與正常代謝者相比,對paroxetine、fluvoxamine、venlafaxine和vortioxetine的藥物暴露或母體與代謝物比率顯著增加(見表S1)。增加的藥物暴露會增加劑量/濃度依賴性副作用的風險(37)。為了可能預防不良反應,根據表現型組間藥代動力學參數差異的劑量推算,建議fluvoxamine劑量減少30%(27)。然而,考慮到目前可用的劑型,減少fluvoxamine劑量30%可能不可行。因此,減少25-50%可能有助於通過限制高藥物暴露來預防不良事件。」
-
這些指引適用於:
- 成人患者
- 兒科考量:「Citalopram、escitalopram和sertraline在兒童中有最多的藥物基因體學數據支持潛在的基因型指導處方變更。基於這些證據,這些藥物的建議適用於兒科患者,並與針對該人群的較小藥代動力學研究一致。」.....「其他建議對兒科患者的普遍性需要確立。因此,治療兒童和青少年的臨床醫師應在考慮這些藥物在青少年中的獨特且更有限的證據基礎以及兒科特有的耐受性差異(例如,激活)和疾病特異性反應軌跡時,確定其對年輕患者的適用性。由於CYP2D6、CYP2C19和CYP2B6的活性在幼兒期達到成人水平,可能適合將與CYP2D6、CYP2C19和CYP2B6相關的抗抑鬱藥的基因型指導建議推廣至青少年或可能更年幼的兒童,並進行密切監測。」
-
下載並閱讀:
表1:基於CYP2D6 表現型的fluvoxamine劑量建議
改編自指引的表1和表2b。
| 表現型 | 活性分數 範圍 |
活性分數 | 範例 | 影響 | 治療 建議 |
建議的 分類a |
考量 |
|---|---|---|---|---|---|---|---|
| CYP2D6 超快速代謝型(Ultrarapid metabolizer) | >2.25 | >2.25 | *1/*1xN, *1/*2xN, *2/*2xNc | 無CYP2D6 超快速代謝型(Ultrarapid metabolizer)的數據可用。 | 無建議由於缺乏證據。 | 無建議 | |
| CYP2D6 正常代謝型(Normal metabolizer) | 1.25 <= x <= 2.25 | 1.25 1.5 1.75 2.0 2.25 |
*1/*10 *1/*41, *1/*9 *10/*41x3 *1/*1, *1/*2 *2x2/*10 |
正常代謝。 | 以建議的起始劑量開始治療。 | 強烈建議 | |
| CYP2D6 中間代謝型(Intermediate metabolizer) | 0 < x < 1.25 | 0.25 0.5 0.75 1.0 |
*4/*10 *4/*41, *10/*10 *10/*41 *41/*41, *1/*5 |
與CYP2D6 正常代謝型(Normal metabolizer)相比,fluvoxamine代謝為活性較低的化合物減少。較高的血漿濃度可能增加副作用的概率。 | 以建議的起始劑量開始治療。 | 中等建議 | |
| CYP2D6 弱代謝型(Poor metabolizer) | 0 | 0 | *3/*4, *4/*4, *5/*5, *5/*6 | 與CYP2D6 正常代謝型(Normal metabolizer)相比,fluvoxamine代謝為活性較低的化合物大幅減少。較高的血漿濃度可能增加副作用的概率。 | 考慮將起始劑量降低25-50%並採用較慢的滴定方案,或考慮選擇一種臨床上合適的替代抗抑鬱藥,其代謝不主要依賴於CYP2D6。 | 可選建議 | 藥物交互作用和其他患者特徵(例如,年齡、腎功能、肝功能)應在調整劑量或選擇替代療法時考慮。 |
| CYP2D6 無法判定 | n/a | 攜帶一個或兩個未知或功能尚未確定 等位基因的個體 | *1/*22, *1/*25, *22/*25 | 無建議 | 無建議 |
a 評分方案詳見補充材料。
2019年10月更新
CYP2D6基因型到表現型翻譯變更:截至2019年8月,CYP2D6基因型到表現型翻譯在指引(即CPIC和DPWG)和臨床基因檢測實驗室之間存在一些不一致。CPIC最近進行了一項修改的Delphi項目,以獲得國際CYP2D6專家小組對統一系統的共識,用於將CYP2D6基因型翻譯為表現型 更多信息。對CPIC先前系統的修改包括將CYP2D6*10 等位基因的活性分數計算值從0.5降至0.25,並將活性分數為1的表現型分配從正常代謝型(Normal metabolizer)更改為中間代謝型(Intermediate metabolizer)(所有先前和新表現型分組的表格)。
因此,CYP2D6 等位基因功能表、CYP2D6基因型到表現型表中已進行以下更改(請參閱下表):
- 雙倍型導致活性分數為1的變更從CYP2D6 正常代謝型(Normal metabolizer)到CYP2D6 中間代謝型(Intermediate metabolizer)分配。
- 對本指引建議的影響:由於本指引中的建議在CYP2D6正常和中間代謝型(Intermediate metabolizer)之間沒有差異,因此目前已發表的正常和中間代謝型(Intermediate metabolizer)的建議將保持不變。
- 所有包含CYP2D6*10 等位基因的雙倍型的活性分數已相應更新(活性分數更改為反映*10的較低值0.25)。在共識項目之前,重複功能正常 等位基因與*10 等位基因的組合導致活性分數為2.5,這轉化為超快速代謝型(Ultrarapid metabolizer)。CYP2D6*10的較低值0.25導致這些等位基因組合的活性分數為2.25,根據共識項目轉化為正常代謝型(Normal metabolizer)。請參閱所有先前和新表現型分組的表格。
- 對本指引建議的影響:本指引的作者目前正在審查受影響的活性分數(AS為2.25)的證據,並將相應更新此網頁和相關表格。
2015年8月
2015年5月在線提前發表
- 關於選擇性血清素再回收抑制劑(SSRI)劑量中使用藥物基因組學測試的指引已由臨床藥物基因體學實施聯盟(CPIC)發表於《Clinical Pharmacology and Therapeutics》期刊。
- 2015年選擇性血清素再回收抑制劑(SSRI)劑量指引摘錄(針對fluvoxamine和CYP2D6):
- 「缺乏描述CYP2D6超快速代謝對fluvoxamine治療影響的數據;因此未提供劑量建議……然而,由於缺乏描述CYP2D6 超快速代謝型(Ultrarapid metabolizer)狀態如何影響fluvoxamine治療的數據,選擇一種不被CYP2D6廣泛代謝的替代SSRI可能是合理的。」
- 「在給予相似劑量時,CYP2D6 弱代謝型(Poor metabolizer)者對fluvoxamine的藥物暴露顯著高於廣泛代謝型(Extensive metabolizer)者……美國食品藥品監督管理局(FDA)指出,已知CYP2D6活性降低的患者應謹慎使用fluvoxamine。為了可能預防不良反應,應考慮為弱代謝型(Poor metabolizer)者選擇一種不被CYP2D6廣泛代謝的替代SSRI。」
- 「指引中的建議主要適用於基於基因檢測的行動;藥物相互作用和其他臨床因素可能對SSRI的處方決策有重大影響,應在開始藥物治療前考慮。」
- 「描述CYP2D6或CYP2C19基因型與SSRI系統暴露或穩定狀態(Steady state)血漿濃度之間關係的數據在兒科患者中稀缺。由於CYP2D6活性在幼兒期已完全成熟,可能適合將這些建議推廣至青少年或可能更年幼的兒童,並進行密切監測。」
- 下載並閱讀:
表1:基於CYP2D6 表現型的fluvoxamine劑量建議:
改編自2015年指引手稿的表1和表2b。
| 可能表現型 | 活性分數 | 基因型 | CYP2D6 雙倍型範例 | 對fluvoxamine代謝的影響 | 治療建議 | 建議等級 a |
|---|---|---|---|---|---|---|
| 超快速代謝型(Ultrarapid metabolizer)(約1-2%的患者)b | > 2.0 | 攜帶功能性等位基因重複的個體 | *1/*1xN, *1/*2xN, *2/*2xN c | 無CYP2D6 超快速代謝型(Ultrarapid metabolizer)的數據可用。 | 無建議由於缺乏證據。 d | 可選建議 |
| 廣泛代謝型(Extensive metabolizer)(約77-92%的患者) | 2.0-1.0 e | 攜帶兩個功能正常 等位基因或兩個功能減弱 等位基因或一個功能正常和一個功能缺失 等位基因或一個功能正常和一個功能減弱 等位基因的個體 | *1/*1, *1/*2, *1/*4, *1/*5, *1/*9, *1/*41, *2/*2,*41/*41 | 正常代謝 | 以建議的起始劑量開始治療。 | 強烈建議 |
| 中間代謝型(Intermediate metabolizer)(約2-11%的患者) | 0.5 | 攜帶一個功能減弱和一個功能缺失 等位基因的個體 | *4/*10,*4/*41, *5/*9 | 與廣泛代謝型(Extensive metabolizer)相比,代謝減少。較高的血漿濃度可能增加副作用的概率。 | 以建議的起始劑量開始治療。 | 中等建議 |
| 弱代謝型(Poor metabolizer)(約5-10%的患者) | 0 | 僅攜帶功能缺失al 等位基因的個體 | *3/*4,*4/*4, *5/*5, *5/*6 | 與廣泛代謝型(Extensive metabolizer)相比,代謝大幅減少。較高的血漿濃度可能增加副作用的概率。 | 考慮將建議的起始劑量減少25-50% f,並根據反應進行滴定,或使用不被CYP2D6代謝的替代藥物。 g | 可選建議 |
a 評分方案詳見補充。
b CYP2D6代謝者狀態頻率基於白人數據,可能與其他族裔不同。請參閱補充說明以獲取不同主要種族/族裔群體中觀察特定雙倍型的機會的信息。
c 其中xN代表CYP2D6基因拷貝數。對於具有CYP2D6重複或多重的個體,請參閱補充數據以獲取有關如何將雙倍型轉換為表現型的信息。
d 缺乏描述CYP2D6超快速代謝對fluvoxamine治療影響的數據;因此未提供CYP2D6 超快速代謝型(Ultrarapid metabolizer)者使用fluvoxamine的劑量建議。然而,由於缺乏描述CYP2D6 超快速代謝型(Ultrarapid metabolizer)狀態如何影響fluvoxamine治療的數據,選擇一種不被CYP2D6廣泛代謝的替代SSRI可能是合理的。
e 活性分數為1.0的患者可能被某些參考實驗室分類為中間代謝型(Intermediate metabolizer)。
f 根據表現型組間藥代動力學參數差異的劑量推算,建議fluvoxamine劑量減少30%(1)。然而,考慮到劑型,減少30%的劑量可能不可行,因此應考慮將fluvoxamine的起始劑量減少25-50%。
g 藥物交互作用和其他患者特徵(例如,年齡、腎功能、肝功能)應在選擇替代療法時考慮。
fluvoxamine 途徑, 藥物動力學
概括
圖示為 fluvoxamine 在肝臟中的代謝過程。
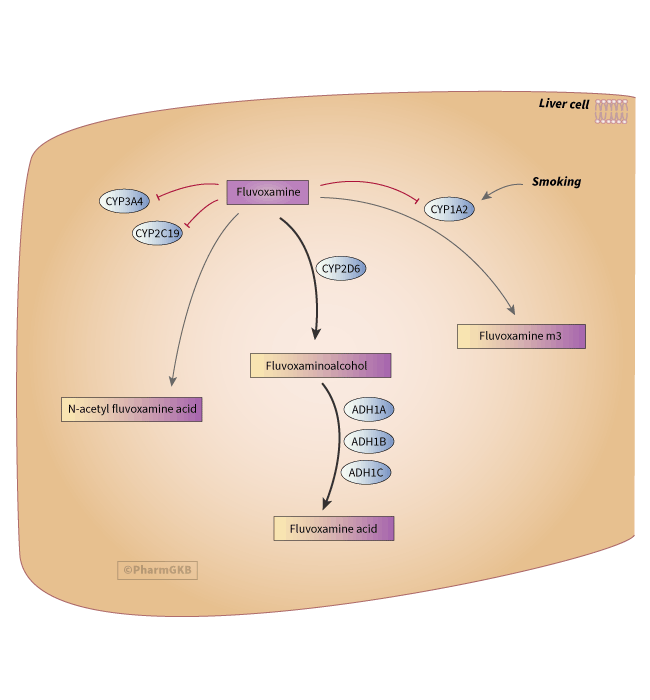
描述
背景
fluvoxamine 是一種 SSRI,用於治療抑鬱症、強迫症、焦慮症和飲食失調 [Article:7988100]。在治療抑鬱症方面,其療效與三環類抗抑鬱藥相當,但抗膽鹼副作用和心血管副作用的發生率較低 [Article:7988100]。最常見的副作用為噁心和胃腸道反應 [Article:7988100]。
代謝
fluvoxamine 在肝臟中被廣泛代謝,尿液中檢測到的母藥物少於4% [Article:11791895]。已在尿液中識別出十一種代謝物,並有九種在文獻中以編號結構記錄 [Article:7988100],但在 PubChem 中發現的結構較少。主要代謝物中沒有顯示出藥理活性 [Article:11791895]。 主要代謝物 fluvoxamine acid(在 PMID 中也稱為 m1: 7988100)約占 fluvoxamine 尿液代謝物的30-60% [Article:11791895]。它是通過 CYP2D6 對甲氧基進行氧化去甲基化的兩步過程生成的,形成 fluvoxaminoalcohol 中間體,隨後由醇脫氫酶(由 ADH1A、ADH1B 和 ADH1C 編碼)生成 fluvoxamine acid [Articles:11791895,17484519]。體外實驗使用微粒體、表達的 CYP 蛋白和 CYP- 特異性抑制劑表明 CYP2D6 是唯一可能執行此過程的 CYP 酶 [Article:17484519]。大約20-40%的 fluvoxamine 尿液代謝物涉及 CYP1A2 對氨基團的取代或去除 [Article:11791895]。去除氨基支鏈後形成代謝物 m3 [Article:7988100]。對兩側鏈的修飾生成代謝物 m4 n-acetyl fluvoxamine acid [Article:7988100]。 在 CYP2D6 PM 中,fluvoxamine 的清除率顯著低於 CYP2D6 EM [Article:8823236]。吸煙增加了 fluvoxamine 在 CYP2D6 PM(n=1)中的清除率,使其與吸煙和不吸煙的 CYP2D6 EM 相當 [Article:8823236]。吸煙誘導 CYP1A2。
Clozapine 途徑, 藥物動力學
概括
肝臟中 clozapine 代謝與運輸之示意圖
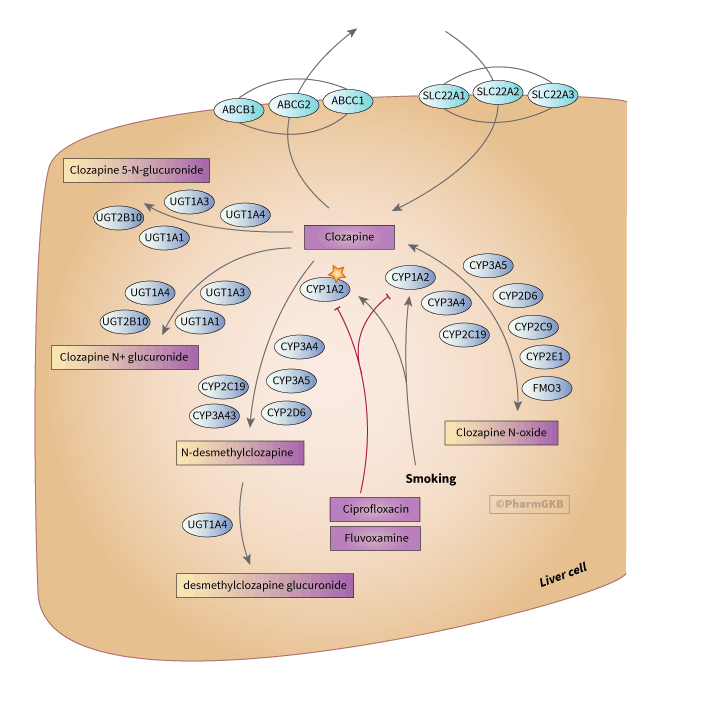
描述
背景
Clozapine是一種非典型抗精神病藥物,也是治療難治性精神分裂症的金標準(TRS),即至少有兩種傳統抗精神病藥物無效的患者[Article:27932669]。指導方針僅允許在TRS中開立Clozapine的原因是其罕見但潛在致命的Clozapine誘導的顆粒細胞缺乏症(CIA)[Article:27932669]。FDA 仿單還列出了黑框警告,指出觀察到與劑量相關的副作用,包括直立性低血壓、心動過緩、暈厥和癲癇發作,並警告老年人有嚴重心臟副作用和死亡率增加的風險(請參見藥物仿單)。其他常見副作用包括鎮靜(大多為暫時性)和代謝性副作用,如體重增加。雖然所需劑量在患者之間可能有很大差異(約150 – 1000mg/天),Clozapine的治療範圍較窄,建議進行治療藥物監測(TDM):Clozapine的血清濃度低於250 ng/mL與復發相關,而高於750 ng/mL則與中毒風險增加相關[Article:27932669。目前尚無針對影響Clozapine代謝的變異體的已發表藥物基因組學(PGx)指導方針,FDA 仿單對CYP2D6 弱代謝型(Poor metabolizer)和潛在的藥物交互作用與同樣CYP450酶代謝的藥物的相互作用提出警告。這個摘要檢視了藥物動力學(PK)Clozapine的代謝及其相關候選基因,並討論了其變異的影響。雖然Clozapine作用於大量受體,可能多達39種不同的受體PDSP Ki數據庫],但藥效學(PD)Clozapine的影響不在本文範疇內,除非PD效應是PK基因的結果。
代謝
Clozapine經過廣泛的肝臟代謝,主要途徑為去甲基化至n-desmethylclozapine和氧化至clozapine n-oxide(如上圖所示)[Article:7891353。體外實驗表明,CYP3A4約占Clozapine清除的70%,CYP1A2約占15%,而CYP2C19、CYP2C8和[[KEEP_358FDA2f]]各占5%或更少[Article:27320963]。對Clozapine代謝的體外研究表明,CYP3A4和CYP1A2是主要負責去甲基化的酶,而CYP2D6的作用非常小[Article:7891353][Article:18809730]。幾種酶能夠在體外生成Clozapine的n-氧化物代謝物(CYP1A2、CYP2E1、CYP2C9、CYP3A4、CYP2D6、FMO3、CYP2C19),但在體內CYP1A2被認為是主要催化劑[Article:7891353][Article:18809730][Article:27932669][Article:23297297]。體內研究表明CYP3A5和CYP3A43也可能發揮作用[Article:28340122][Article:25150845]。n-desmethylclozapine是一種活性代謝物,能夠通過多巴胺D2和D3受體發揮作用[Article:19483482],組胺受體[Article:21912901],毒蕈鹼M1[Article:25859763],以及血清素受體[Article:17583355]。此外,Clozapine與n-desmethylclozapine的比率是預測精神分裂症患者工作記憶表現的強烈建議指標;較低的比率與更好的工作記憶和執行功能相關[Article:25859763][Article:29024893]。clozapine n-oxide被認為是無活性的,可能會被代謝回Clozapine [Article:7891353][Article:16805946][Article:28664816]。在接受Clozapine治療的患者尿液中已識別出額外的代謝物,但這些代謝物的臨床意義尚不清楚[Article:9833598]。
雖然在CYP1A2中存在功能變異體,但對這些變異體的影響尚無共識,特別是與Clozapine相關[Article:15206669][Article:15949157][Article:16044115][Article:21481946][Article:25090458]。吸煙者的Clozapine血清濃度低於非吸煙者,這是由於CYP1A2的活化[Article:11476124]。吸煙行為的變化可以顯著改變Clozapine的代謝;在戒煙後,Clozapine的暴露增加超過50%[Article:11763003]。強烈建議的CYP1A2抑制劑,如抗生素ciprofloxacin,也可以顯著增加Clozapine的暴露,並且病例研究報告了致命的相互作用[Article:27872784]。此外,在感染期間,促炎細胞因子可以下調CYP1A2的表達,這可能會加劇這一效應[Article:25519488]。抗抑鬱藥fluvoxamine也是CYP1A2的抑制劑,並且可以增加Clozapine的血漿濃度。這一PK效應可以用作調節Clozapine劑量的機制;減少Clozapine的劑量以嘗試減少副作用,同時保持療效[Article:10982203]。
總體而言,劑量、性別、年齡和吸煙約占血清Clozapine變異性的50%,其餘50%可能由藥物代謝酶的基因變異和共同用藥組成[Article:9798077][Article:2813665]。男性的血清Clozapine濃度低於女性,儘管女性接受的劑量較低[Article:2813665][Article:8195452][Article:9798077]。吸煙者的血漿Clozapine濃度約低20-30%相比非吸煙者[Article:2813665][Article:8195452][Article:9798077]。年長患者(45歲以上)的濃度顯著高於年輕患者(18-26歲)[Article:2813665]。一項針對接受抗精神病藥物的孕婦的小型研究發現,雖然對Clozapine(n=4)的血漿濃度下降並不顯著,但對其他藥物(阿立哌唑n=14和奎etiapine n=35)則顯著,作者推測在懷孕期間CYP3A4的水平會增加,這可能會影響抗精神病藥物的PK[Article:28643331]。這與懷孕期間行為的改變(如咖啡因使用和吸煙的變化)相結合,可能對接受Clozapine的女性進行監測非常重要。
運輸
關於Clozapine運輸的實驗涉及多種細胞類型,尚未提供關於哪些運輸蛋白參與進入或排出的統一圖景。以下是分散的證據,所有合理的候選者在圖1中顯示:
Clozapine有效地穿越血腦屏障並進入大腦,如PET對C11的成像所示仿單的Clozapine [Article:26225256]。然而,這項研究使用了靜脈注射Clozapine,而大多數患者是口服給藥,且負責的運輸蛋白未被確定。
對於口服Clozapine,腸道的運輸非常重要。有證據表明,Clozapine在腸道的代謝與在肝臟的代謝不同,因為葡萄柚汁(腸道CYP3A4的抑制劑,但不是肝臟CYP3A4的抑制劑)不影響血漿Clozapine濃度[Article:11791889][Article:11816871]。
肝臟中的攝取可能由SLC22A1催化,因為Clozapine能夠在體外抑制SLC22A1對模型底物(MPP+)的運輸[Article:22806583]。在同樣的體外實驗中,Clozapine也與SLC22A2和SLC22A3相互作用[Article:22806583]。
Clozapine與ABCB1編碼的運輸蛋白(也稱為PgP)之間的關係在文獻中存在矛盾。早期的體外實驗使用CaCo2細胞(腸道細胞系)顯示Clozapine和n-desmethylclozapine是ABCB1的低親和力底物,並且是ABCB1-介導的塔尼洛洛運輸的抑制劑[Article:15285840]。然而,儘管ABCB1變異體與不同的PK或結果相關[Article:19593168][Article:28919802],最近的文章報導Clozapine不是ABCB1的底物或有效抑制劑[Article:21843066]。
Clozapine在體外抑制ABCG2對米托坦的運輸[Article:18834354]。此外,ABCG2運輸蛋白的變異體與Clozapine的暴露相關,而ABCC1的變異體與結果相關,但尚未明確顯示Clozapine是兩者的底物(在PGx部分進一步討論)[Article:27932669][Article:28919802]。
藥物交互作用,DDIs
有關Clozapine 藥物交互作用的出版物很多,但大多數研究規模很小或為個案報告,且很少考慮PGx變異體是否影響相互作用(在[Article:17214606]中回顧)。 FDA 仿單對Clozapine的分類將DDI分為四種類型,並對每種類型提出建議:
- 強烈建議 CYP1A2抑制劑,例如fluvoxamine和ciprofloxacin,建議將Clozapine劑量減少至三分之一。
- 中等建議或弱CYP1A2抑制劑,如口服避孕藥或咖啡因,建議監測不良反應並考慮減少劑量。
- CYP2D6或CYP3A4抑制劑,如氟西汀,建議監測不良反應並考慮減少劑量。
- 強烈建議 CYP3A4誘導劑不建議使用,例如卡馬西平、利福平、苯妥英和聖約翰草。 每種類型的DDI研究在下文中討論並在表1中總結:
類型1 DDIs:強烈建議 CYP1A2抑制劑
文獻中有多篇報告顯示Clozapine與ciprofloxacin之間存在嚴重且有時致命的相互作用,支持調整Clozapine劑量的建議[Article:27872784][Article:23346515][Article:22929408][Article:19067475][Article:18354073][Article:17624521][Article:17329613][Article:11151749]。還有大量文獻關於fluvoxamine和Clozapine(在[Article:17214606]中回顧):幾篇論文討論了使用fluvoxamine作為輔助藥物以避免高劑量Clozapine;較低的Clozapine劑量減少了包括體重增加[Article:25627831]、葡萄糖/代謝功能障礙[Article:28688742]、和血液毒性[Article:23490199]的副作用發生率。 一項使用人類肝臟微粒體的體外研究調查了與fluvoxamine共同治療時n-desmethylclozapine和clozapine n-oxide的產生。作者得出結論,咖啡因表型在預測CYP1A2-相關的Clozapine-fluvoxamine DDI時並不可靠[Article:29155491]。
類型2 DDIs:中等建議或弱CYP1A2抑制劑
咖啡因攝入是血清Clozapine水平的重要預測因子[Article:23104241]。各種案例研究顯示,咖啡因攝入可以影響Clozapine的血漿水平和Clozapine的療效[Article:8601555][Article:17515710]。一項隨機研究交替讓患者飲用常規咖啡或無咖啡因咖啡,顯示咖啡因與血漿Clozapine相關[Article:14725610]。咖啡因攝入也可以來自可樂飲料,其中一天六杯的攝入在一個案例研究中使血漿Clozapine達到毒性水平[Article:22926611]。個體對咖啡因的血漿Clozapine反應的變異受到CYP1A2的影響[Article:14725610]。
有幾個案例報告顯示DDI與Clozapine和口服避孕藥的相互作用[Article:12454563][Article:25890012][Article:24717251]。在現實情況中,監測和考慮劑量調整的建議可能會更複雜,因為開處方的醫生可能不知道患者的避孕藥處方歷史,而患者也可能未意識到分享這些信息的重要性。
類型3 DDIs:CYP2D6或CYP3A4抑制劑
與由CYP2D6代謝的抗抑鬱藥(包括舍曲林、帕羅西汀和氟西汀)共同治療的Clozapine報導導致血漿Clozapine增加20-40%(在[Article:24494611]中回顧)。雖然Centorrino的研究檢查了40名與SSRIs共同治療的患者和40名單獨接受Clozapine的患者,顯示血清Clozapine和n-desmethylclozapine增加,但血清濃度的標準差非常大(例如,與帕羅西汀共同治療的患者的平均Clozapine濃度為417ng/ml,標準差為373ng/ml)[Article:8633698]。另一項對14名接受帕羅西汀和Clozapine的患者的研究顯示Clozapine或主要代謝物沒有變化[Article:9928923][Article:9472836]。在一個個案報告中,一名接受Clozapine和帕羅西汀的患者血清Clozapine出現毒性增加{Joos, 1997 #104}。而一項對9名與帕羅西汀共同治療的患者和8名與舍曲林共同治療的患者的研究發現,僅在帕羅西汀組中Clozapine和N-去甲基Clozapine增加[Article:11147928。然而,由於帕羅西汀也由CYP1A2和CYP3A4代謝,因此這一關係可能並不完全由CYP2D6的抑制引起,請參見帕羅西汀途徑]。 在一項體外微粒體實驗中,CYP2D6的抑制劑奎尼丁和右美沙芬影響Clozapine的代謝,並減少了次要代謝產物的生成,如HPLC所示,但未改變n-desmethylclozapine或clozapine n-oxide的量[Article:1545398]。
類型4 DDIs:強烈建議 CYP3A4誘導劑
雖然不建議共同開處方Clozapine和強烈建議 CYP3A4誘導劑,但仿單列舉了如苯妥英和卡馬西平的例子,但不包括奧卡西平。關於奧卡西平的案例報告表明,它可能特別容易通過Clozapine與CYP3A4和CYP1A2相互作用[Article:28255440][Article:26711482]。
其他DDIs
Clozapine的副作用之一是胃食道逆流病,GERD [Article:28291036]。GERD的治療是氫離子幫浦阻斷劑(PPI)(PPIs),其中大多數由CYP2C19代謝。DDI與Clozapine和PPIs之間有幾種可能的相互作用路徑;CYP1A2的誘導、FMO3的誘導,以及CYP2C19的競爭性抑制。這些相互作用中的任何一種或全部都可能導致n-desmethylclozapine和clozapine n-oxide的增加,並增加血液毒性的風險[Article:28291036]。clozapine n-oxide在體外抑制CYP2C19介導的-美芬妥因的代謝[Article:27853934]。因此,clozapine n-oxide可能會抑制PPIs的代謝。
中等建議或弱CYP1A2或CYP3A4誘導劑也在藥物仿單中討論,建議在觀察到療效不足時增加劑量。
藥物仿單還警告在已經有長QT風險的人群中使用Clozapine,並提到許多可能增加長QT綜合症風險的藥物。列出的幾種藥物也被禁用作CYP3A4的抑制劑,例如紅霉素或CYP2D6的抑制劑,例如奎尼丁、氯丙嗪或CYP1A2的抑制劑,例如胺碘酮。
藥物基因體學
雖然大多數有關Clozapine的PGx研究已檢查PD候選基因,但本文專注於PK候選基因及其對PK或PD/臨床結果的影響(在表2中總結)。候選基因CYP1A2、CYP3A4、CYP2C19、CYP2D6和ABCB1都是知名的藥物基因。此外,還有詳細描述每個PGx關係的論文,這些論文合併到變異註釋標籤下的互動表中。
CYP1A2
目前尚無與CYP1A2代謝相關的編碼序列變異,這可能是由於在大多數人群中的頻率非常低(在{Thorn, 2012 #63}中回顧,並在CYP1A2 VIP摘要中總結)。最常研究的變異位於上游啟動子區域:CYP1A2:(-163)C>A (rs762551),CYP1A2:(-3860)G>A (rs2069514)和CYP1A2:(-729)C>T (rs12720461),這些變異形成了單倍型 *1F、*1C、*1D和*1K [Article:21989077]。由於歷史上對於-164(rs762551)哪個變異代表*1F的混淆,具體變異在報告中給出。單倍型 *1F被認為是一種可誘導的變異,具有高活性,單倍型 *1C、*1D和*1K被認為是低活性[Article:21989077]。
在接受Clozapine的患者中,CYP1A2*1F與治療不反應相關,需要更高劑量[Article:28356835][Article:15206669][Article:16044115][Article:15949157]。CYP1A2 *1F(rs762551 AA)的純合子吸煙者的血漿Clozapine較低,代謝物濃度較高,清除速度較快,與非吸煙者相比。在一項針對4名對Clozapine無反應的重度吸煙者(每天30支或更多香煙)的案例研究中,發現他們都是同型合子(Homozygous) *1F(描述為-164C > A,均為AA)[Article:15206669]。所有患者在Clozapine血漿水平提高至治療閾值以上後,無論是通過將Clozapine劑量提高至非常高的值,還是通過引入fluvoxamine,均經歷了臨床狀態的顯著改善[Article:15206669]。另一個案例報告中,兩名患者戒煙後出現嚴重不良反應:一名吸煙超過40支/天的患者突然戒煙,血漿Clozapine極高且出現不良反應;另一名報告每天吸煙3或4支的患者住院且不被允許吸煙,並有多種共同用藥。兩名患者均為同型合子(Homozygous) CYP1A2*1F,具體定義為-164AA[Article:16044115]。此外,對58名接受Clozapine的精神分裂症患者的研究發現,吸煙者的濃度劑量比(C/D)比CYP1A2*1F(AA)低,但這並不顯著[Article:15949157]。非吸煙者的*1F雜合子或純合子有較高的C/D比率。吸煙者被描述為每天至少吸煙15支,但範圍未報告[Article:15949157][Article:12618594]。
接受Clozapine的患者在劑量較低(< 300 mg/d)時,癲癇風險較低,而在維持劑量較高(>或= 600 mg/d)時,癲癇風險較高[Article:7991106]。同型合子(Homozygous) CYP1A2*1F基因型與接受Clozapine的患者癲癇風險增加相關(n=108)[Article:23601795],這可能反映了CIA*1F與高劑量的關聯。
表達低的等位基因的CYP1A2患者更可能出現與血漿Clozapine增加、代謝物減少和清除減少相關的不良事件。一項針對三名對Clozapine不耐受且出現遲發性運動障礙(TD)的患者的案例研究,這些患者在低/正常劑量下未使用其他混雜藥物,發現他們的基因型為CYP1A2*1C(2 異型合子(Heterozygous)、1 同型合子(Homozygous)且均無*1F),這是一種低表達等位基因。這些患者淋巴細胞中的CYP1A2-mRNA表達水平低於對照組的1/30[Article:21481946]。另一項針對精神分裂症住院患者的案例系列中,有部分患者接受Clozapine(209名患者中有18名),發現低活性CYP1A2(無誘導劑CBZ、VPA或吸煙,CYP1A2*1D delT或CYP1A2*1F C)與PSP-P和CGI-2分數測量的臨床結果下降相關[Article:25090458]。
一般來說,Clozapine和BMI的PGx研究以及與代謝綜合徵相關的副作用主要集中在PD基因上,並未包括PK基因變異的測量。對於代謝副作用是否與劑量相關存在一些意見分歧[Article:26364648]。一項小型研究(n=17)顯示,低活性變異體CYP1A2 *1C和*1D與較高的血清Clozapine濃度相關,並且在給定劑量的Clozapine上增加了發展胰島素和脂質升高及胰島素抵抗的風險[Article:17503978]。
CYP3A
低CYP3A4表達(以白細胞mRNA水平測量)與高血清Clozapine相關[Article:28340122]。在這些低表達的CYP3A4中,CYP3A5的表達,即CYP3A5*1,導致與CYP3A5*3純合子相比,血清Clozapine減少,且後者未表達功能性CYP3A5 [Article:28340122]。另一項研究顯示,CYP3A5對CYP3A4的影響僅在活動降低的患者中有所貢獻[Article:19593168]。
CYP3A43是CYP3A位點中的另一個基因[Article:20019904],儘管其在Clozapine 途徑中的具體參與尚不清楚,但變異rs680055和rs472660與對Clozapine的反應增加相關[Article:25150845]。
CYP2D6
早期對接受Clozapine的患者的研究顯示,PM和EM基因型與反應之間沒有關聯[Article:7640149]。其他幾項研究得出結論,CYP2D6變異體與Clozapine/n-desmethylclozapine比率或代謝效應並無顯著關聯[Article:7640149][Article:17503978][Article:19593168] [Article:8148222]。FDA藥物仿單未提供支持包含有關CYP2D6變異的文字的參考。
CYP2C19
一項對接受Clozapine的患者的研究顯示,CYP2C19*17/*17基因型,被認為是超快速代謝型(Ultrarapid metabolizer),血清n-desmethylclozapine較高,與糖尿病的較低發病率相關,並且與其精神分裂症症狀的改善增加相關,與CYP2C19*1/*1相比[Article:28664816]。在另一項研究中,弱代謝型(Poor metabolizer)基因型CYP2C19*2/*2與較高的血清Clozapine相關[Article:19593168]。CYP2C19*17/*17與全體隊列中的血清Clozapine或n-desmethylclozapine的顯著變化無關,但在未接受fluvoxamine的小組中卻顯著[Article:19593168]。在一項對91名患者的大型研究中,無論是*2還是*17均未與血清Clozapine的變化相關[Article:23297297]。
運輸蛋白變異
有幾項研究檢查了運輸蛋白中的變異,儘管尚未重複。ABCB1變異rs1045642 AA基因型(3435G>A)與血漿Clozapine增加相關[Article:19593168]。ABCB1的相同基因型rs1045642也與顆粒細胞缺乏症和嗜中性白血球低下症相關[Article:27168101],但另一項研究發現,當該基因型是單倍型的ABCB1的一部分時,可能對顆粒細胞缺乏症具有保護作用[Article:27043126]。一般來說,Clozapine誘導的顆粒細胞缺乏症不被認為是劑量依賴的,但特別是在白細胞中的藥物濃度增加可能是一個因素,並可能受到基因變異的影響。變異rs1045642G在ABCB1和rs212090T在ABCC1與男性但非女性接受Clozapine的體重增加和高血壓風險增加相關[Article:28919802]。
ABCB1 rs7787082 G和rs10248420 A 等位基因在對Clozapine無反應的患者中更常見[Article:22722500]。變異rs2231142在ABCG2中與Clozapine的劑量調整血清谷濃度(Ctrough)增加相關[Article:27932669]。
結論
我們簡要總結了參與Clozapine代謝的候選基因及其在DDI和PGx反應中的作用。Clozapine的PK變異性仍有很大一部分未得到充分解釋。來自DDI的體外和體內實驗的證據並不完全一致[Article:8633698][Article:9928923][Article:9472836][Article:29024893]。這可能有許多原因,包括不同細胞類型中的代謝差異、考慮不同的代謝物以及PGx影響。體外實驗是使用表達的蛋白質或肝臟微粒體進行的,而體內研究可能涉及許多細胞和器官系統。體外系統不允許對CYP1A2或CYP3A4的轉錄調節,這可能是DDI的一個重要部分。 這兩類DDI研究也主要集中在母藥及其兩個主要血清代謝物n-desmethylclozapine和clozapine n-oxide上,對於次要代謝物和途徑在n-desmethylclozapine下游的相關性知之甚少。很少有研究考慮PGx變異對DDI的影響。
目前,PGx證據非常分散,且很少有關聯被重複,但CYP1A2、CYP2C19和CYP3A家族基因都是良好的候選者。CYP2D6變異對Clozapine的PGx影響缺乏證據,儘管DDI的研究顯示CYP2D6參與Clozapine的PK,並且可能在生成次要代謝物中發揮作用[Article:7891353]。 [Article:11147928][Article:8195452][Article:28291036]。需要更大規模的體內研究,包括量化環境影響(如咖啡因和吸煙)以及多個PGx候選基因的機制。雖然一項小型研究得出吸煙對Clozapine/n-desmethylclozapine血漿濃度的影響與每天吸煙的香煙數量無關(>20、11-20、6-10、<=5),但該研究僅檢查了45名吸煙者[Article:19593168]。最近一項將咖啡因代謝和863名健康個體(包括385名吸煙者)的吸煙數據結合的研究,模型化Clozapine的代謝,顯示香煙數量與清除率相關。該模型能夠預測Clozapine的清除率,與已發表的每日香煙數量範圍(>20、11-20、6-10、<=5)一致[Article:22258279]。生成這樣的模型的困難在於考慮咖啡消費可能對CYP1A2表達的誘導影響[Article:18157525]。被動吸煙也可能需要考慮,因為有證據表明它影響了苯乙烯的代謝,苯乙烯是CYP1A2的另一種底物。被動吸煙者的苯乙烯代謝介於吸煙者和非吸煙者之間[Article:9712458]。此外,患者的居住狀況變化應在任何預測模型中考慮,因為從家庭護理、醫院急診護理和住院設施護理的變化可能影響吸煙行為。預測護理安排變化的影響大小的能力可能有助於避免不良事件。還需考慮其他誘導劑/抑制劑,包括環境毒素(如果患者處於不穩定的居住情況下可能會遇到)、處方藥、濫用藥物、替代藥物、酒精和飲食。有證據表明,炎症誘導的下調CPY1A2和CYP3A4導致從EM基因型轉變為PM表現型 [Article:25519488]。
需要更多有關CYP3A5和CYP3A43的貢獻數據。雖然基因變異似乎對CYP3A4的影響有限,但有些變異對CYP3A5和CYP3A43的影響很大[Article:28340122][Article:25150845]。此外,運輸蛋白變異在肝臟和血腦屏障的影響需要更一致的檢查。將所有這些因素結合到演算法中,連同PD候選基因的因素,是最終目標。
選擇性血清素再回收抑制劑(SSRI) 途徑, 藥效學
概括
參與血清素合成、釋放、再攝取及介導抗憂鬱劑 選擇性血清素再回收抑制劑(SSRI)在人類大腦中作用的基因。
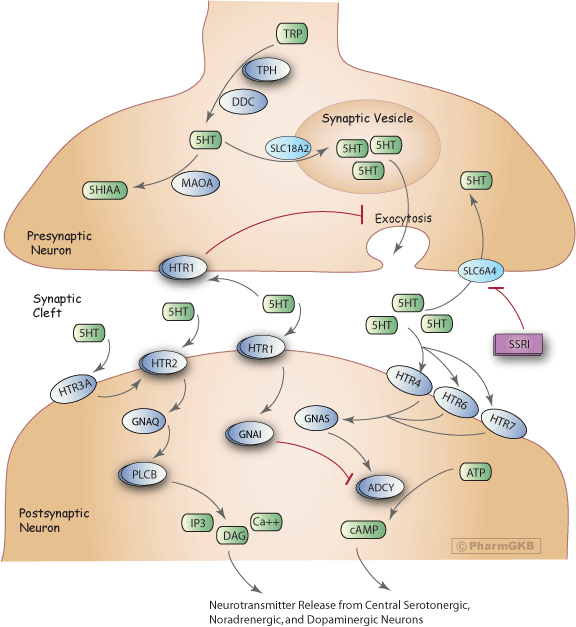
描述
血清素(5-羥色胺,5-HT)是一種神經傳導物質,影響多個過程,包括自主功能、運動活動、荷爾蒙分泌、認知以及與情感、情緒和獎勵相關的複雜過程[Article:17046718]。
在血清素能神經元的末端軸突中,自由色氨酸(TRP)被轉化為5-HT[Article:11590474]。5-HT的合成是一個由色氨酸羥化酶(TPH)和芳香族脫羧酶(DDC)催化的兩步驟過程。TPH是限速酶,存在兩種同工型TPH1和TPH2。在神經組織中,TPH2同工型是主要形式[Articles:12511643, 16581041]。5-HT的攝取進入突觸前儲存囊泡是由囊泡單胺轉運蛋白(SLC18A2)介導的。該轉運蛋白利用囊泡膜上的質子梯度將血清素積累到突觸囊泡中[Article:9665836]。未儲存於囊泡中的5-HT則被單胺氧化酶A(MAOA)降解為5-羥基吲哚乙酸(5-HIAA)。
動作電位刺激突觸前囊泡中血清素的鈣依賴性外排,釋放至突觸間隙,並與突觸後和突觸前受體相互作用。在突觸前側,5-HT激活5-羥色胺(血清素)受體1A(HTR1A)、B(HTR1B)和D(HTR1D),導致5-HT外排的減弱[Article:11590474]。這一反饋迴路調節突觸間隙中的5-HT濃度,因此影響突觸後膜上各種HTR受體亞類的刺激程度[Article:8480026]。長期使用選擇性血清素再回收抑制劑(SSRI)會使這些反饋迴路失去敏感性,從而削弱其對血清素能神經傳導的調節作用[Article:8221701]。突觸後HTR1受體(HTR1A、HTR1B、HTR1D、HTR1E、HTR1F)與HTR2受體亞型(HTR2A、HTR2C)共同作用,通過激活第二信使級聯來介導效應信號[Article:11590474]。在突觸後,訊息傳遞途徑對HTR1受體亞型的主要信號鏈接是通過Gi/o蛋白α亞單位(GNAI)的耦合來實現的。這一相互作用通過抑制腺苷酸環化酶(ADCY)來降低環狀AMP的形成[Article:16896947]。在與5-HT相互作用後,HTR2受體亞群的主要信號鏈接是通過耦合Gq/11蛋白α(GNAQ)來激活磷脂酶C(PLCB)[Article:16896947]。PLCB催化肌醇-1, 4, 5-三磷酸(IP3)和二酸甘油(DAG)的形成[Article:11916537]。突觸後的離子型HTR3受體是一種陽離子特異性配體門控離子通道,並不激活第二信使系統[Article:18466097]。5-HT與此受體的結合通過鈉的內流和鉀的外流使突觸後膜去極化,這被認為會影響HTR2受體的激活[Article:11590474]。HTR4、HTR6和HTR7主要耦合Gs蛋白α(GNAS),導致腺苷酸環化酶的激活,從而增加環狀AMP的水平[Article:11916537]。進一步的操作多樣性得益於多種HTR的剪接和編輯變異的存在、輔助蛋白和伴護蛋白的可能調節,以及它們在GPCR和配體門控通道層面形成同源或異源聚合物的潛力[Article:18571247]。
所有這些第二信使信號在進一步下游反應中的放大導致通過調節鉀通道、幾種蛋白激酶和其他鈣依賴性信號來介導來自中樞血清素能、去甲腎上腺素能和多巴胺能神經元的神經傳導物質釋放。
慢性使用抗抑鬱劑治療已被報導通常會增加腦源性神經生長因子(BDNF)的表達,這是一種依賴於活動的分泌蛋白,對神經網絡的組織和突觸可塑性至關重要[Articles:18433734, 19403460]。
溶質運輸蛋白家族6(神經傳導物質轉運蛋白,血清素),成員4(SLC6A4)負責終止突觸間隙中5-HT的作用。釋放的血清素通過這種整合膜蛋白被運輸回突觸前末端。SLC6A4是Na+/Cl--依賴性轉運蛋白家族的成員[Article:14642278]。
目前存在四大類抗抑鬱藥物:單胺氧化酶抑制劑、選擇性血清素再回收抑制劑(SSRI)、三環化合物和非典型抗抑鬱藥[Article:8221701]。迄今為止,尚未建立針對這些療法的全面假說來解釋其抗抑鬱作用[Article:12726882]。儘管如此,這些藥物都有一個或多個主要的分子靶點以發揮作用。SSRI的分子靶點是SLC6A4,導致抑制突觸前從突觸間隙中5-HT的再攝取。五種SSRI(fluoxetine、fluvoxamine、paroxetine、sertraline和citalopram)在其藥理特徵上有所不同,導致對特定患者的療效或副作用特徵的差異[Articles:10674711, 19370626, 18308785]。SSRI對5-HT再攝取轉運蛋白具有高親和力,對去甲腎上腺素再攝取轉運蛋白具有低親和力,對神經傳導物質受體則具有非常低的親和力。
目前的抗抑鬱療法需要持續治療2-4週才能有效的觀察結果,這表明血清素能和去甲腎上腺素能神經傳導及下游神經適應(例如BDNF受體訊息傳遞途徑)的適應性變化,而不僅僅是突觸單胺水平的升高,才是其治療效果的原因[PMID:19522734, 19481572]。
幾種基因多態性已與治療SSRI反應及不良反應相關,包括SLC6A4、HTR1A、HTR2A、HTR3B、TPH、BDNF和G蛋白β3亞單位的基因變異[Articles:18466097, 19403460, 15111987, 19558256]。