CPIC 指南註解:celecoxib、flurbiprofen、ibuprofen、lornoxicam 與 CYP2C9 基因
摘要
CPIC 對 celecoxib、flurbiprofen、ibuprofen 和 lornoxicam 的劑量指引建議,對於 CYP2C9 弱代謝型(Poor metabolizer),應以最低建議起始劑量的 25-50% 開始治療;對於 CYP2C9 中間代謝型(Intermediate metabolizer)活性分數為 1 的患者,應以最低建議起始劑量開始治療。請參閱完整指引以獲取更多詳細信息和支持證據。
為特定註釋指定基因型或表現型
註釋
此註釋基於CPIC®指引,針對非類固醇抗炎藥和CYP2C9。
2020年3月
2020年3月線上提前發表。
-
關於CYP2C9和非類固醇抗炎藥的CPIC指引已發表於《Clinical Pharmacology and Therapeutics》期刊。
-
這些指引適用於:
-
兒科患者
-
成人患者
-
-
2020年非類固醇抗炎藥劑量指引摘錄:
-
「有充分證據表明CYP2C9基因型與CYP2C9代謝和血漿NSAID濃度的表型變異性相關,大多數研究是在健康志願者中進行的。」
-
「雖然臨床證據顯示CYP2C9的基因變異與NSAIDs使用增加不良事件的速率之間的關聯性有限,但幾項研究已建立CYP2C9 功能減弱和功能缺失 等位基因與NSAID暴露增加之間的關聯(圖1和圖S2)。由於大多數NSAID不良事件是劑量依賴性的,涉及COX抑制的目標不良事件(19-23),因此合理假設暴露增加會增加不良事件的風險。」
-
「CYP2C9 IM和PM 表現型會通過降低代謝清除率影響NSAIDs的系統血漿濃度,從而延長血漿消除半衰期。因此,治療建議根據NM中的NSAID血漿消除半衰期進行廣泛組織。當有超過兩項研究報告血漿濃度曲線下面積(AUC)時,進行了元分析以估計CYP2C9基因型對藥物暴露的平均影響(圖1和圖S2至S4)。」
-
「Celecoxib, flurbiprofen, ibuprofen, lornoxicam:根據現有證據,AS為1.5的NMs和IMs建議以核准的起始劑量開始治療。儘管代謝輕微減少,AS為1.5的IMs相對於NMs並未顯示出顯著增加的藥物暴露... AS為1的CYP2C9 IMs代謝減少,預期會顯示出延長的藥物半衰期和較高的血漿濃度,這可能增加毒性的可能性。對於AS為1的IMs,建議以最低建議起始劑量開始NSAID治療,並根據臨床效果進行劑量調整,密切監測治療過程中的不良事件,如血壓升高和腎功能障礙。關於ibuprofen的使用,應考慮到雖然單獨的CYP2C9*2 等位基因可能不會導致臨床相關的清除率減少,但其與強烈建議的連鎖可能導致R(-) ibuprofen羥化受損和母藥暴露增加...具有CYP2C9 PM 表現型(AS為0)的個體預期代謝顯著減少,預期會顯示出顯著延長的藥物半衰期和血漿濃度增加,這可能增加毒性的可能性和/或嚴重性...建議以最低建議起始劑量的25-50%開始治療(即50-75%劑量減少),並謹慎調整劑量以達到臨床效果。由於這些患者的藥物半衰期顯著延長,劑量上調不應在達到穩態之前進行,考慮到每種藥物的PM半衰期;當然,因毒性可隨時停止或減少劑量。也可以考慮使用替代療法。這可能包括非主要由CYP2C9代謝的NSAIDs(如aspirin, ketorolac(僅批准短期使用), metamizole, naproxen, sulindac, etoricoxib, parecoxib或valdecoxib),或在體內CYP2C9基因變異顯然不影響藥代動力學參數的藥物,儘管在體外CYP2C9代謝。」
-
「Meloxicam。Meloxicam的半衰期(15-20小時,表S12)比celecoxib和ibuprofen長,因此預期meloxicam代謝受損會導致藥物暴露持續升高。對於AS為1.5的CYP2C9 NMs和IMs的建議類似於短半衰期NSAIDs,包括以標準劑量開始治療,同時使用最低有效劑量以達到治療目標的最短持續時間。對於AS為1的IMs,預期代謝減少和血漿濃度增加,這可能增加毒性的可能性...建議要麼以最低建議起始劑量的50%開始治療,要麼選擇替代療法,這與短半衰期NSAIDs的PMs建議一致(表2)。劑量上調不應在達到穩態之前進行(至少七天),並建議進行仔細監測。CYP2C9 PMs應開具替代療法,因為預期半衰期顯著延長(即,>100小時)」
-
「Piroxicam和tenoxicam。這些藥物的半衰期極長(分別為30-86和60小時),因此在CYP2C9代謝減少的個體中放大了潛在風險,並由於缺乏數據而阻礙了劑量調整策略。因此,IMs(AS為1)和PMs建議接受替代療法。這包括不由CYP2C9代謝或在體內不受CYP2C9基因變異顯著影響的藥物。也可以考慮選擇半衰期短的NSAID(表2)。」
-
「Aceclofenac, aspirin, diclofenac, indomethacin, lumiracoxib, metamizole, nabumetone和naproxen。這些藥物的藥物動力學在體內不受CYP2C9基因變異顯著影響和/或目前沒有足夠的證據提供指導臨床實踐的建議(CPIC建議分類「無建議」;CPIC等級C」
-
「兒科:由於CYP2C9活性在幼兒期已完全成熟,可能適合將這些建議外推至青少年或可能更年幼的兒童,並進行密切監測。最終,仍需在兒科患者中進行更多研究和臨床試驗,以調查CYP2C9基因型與NSAID系統暴露和治療結果之間的關聯。」
-
-
下載並閱讀:
表1:根據CYP2C9 表現型celecoxib, flurbiprofen, ibuprofen, lornoxicam的推薦劑量
改編自2020指引的表1和表2。
| 表現型a | 活性分數 | 基因型 | 基因型示例b | 影響 | 治療建議c | 建議等級d | 其他考量 |
|---|---|---|---|---|---|---|---|
| CYP2C9 正常代謝型(Normal metabolizer) | 2c | 攜帶兩個功能正常等位基因的個體 | *1/*1 | 正常代謝 | 以建議的起始劑量開始治療。根據處方信息,使用最低有效劑量以達到個別患者治療目標的最短持續時間。 | 強烈建議 | |
| CYP2C9 中間代謝型(Intermediate metabolizer)f | 1.5c | 攜帶一個功能正常和一個功能減弱 等位基因的個體 | *1/*2 | 代謝輕微減少 | 以建議的起始劑量開始治療。根據處方信息,使用最低有效劑量以達到個別患者治療目標的最短持續時間。 | 中等建議 | IMs可能有比正常更高的不良事件風險,特別是在有其他影響這些藥物清除因素的個體中,如肝功能受損或高齡。對於攜帶CYP2C9*2 等位基因的個體使用ibuprofen時應特別小心,因為它與CYP2C8*3有連鎖,且ibuprofen也由CYP2C8代謝。 |
| CYP2C9 中間代謝型(Intermediate metabolizer)f | 1c | 攜帶一個功能正常 等位基因加上一個功能缺失 等位基因或兩個功能減弱等位基因的個體 | *1/*3, *2/*2 | 中等建議代謝減少;較高的血漿濃度可能增加毒性的可能性 | 以最低建議起始劑量開始治療。謹慎地將劑量上調至臨床效果或最大建議劑量。根據處方信息,使用最低有效劑量以達到個別患者治療目標的最短持續時間。在治療過程中仔細監測不良事件,如血壓和腎功能。 | 中等建議 | IMs可能有比正常更高的不良事件風險,特別是在有其他影響這些藥物清除因素的個體中,如肝功能受損或高齡。對於攜帶CYP2C9*2 等位基因的個體使用ibuprofen時應特別小心,因為它與CYP2C8*3有連鎖。 |
| CYP2C9 弱代謝型(Poor metabolizer) | 0或0.5c | 攜帶一個功能缺失 等位基因加上一個功能減弱等位基因;或兩個功能缺失等位基因的個體 | *2/*3, *3/*3 | 代謝顯著減少和半衰期延長;較高的血漿濃度可能增加毒性的可能性和/或嚴重性 | 以最低建議起始劑量的25-50%開始治療。謹慎地將劑量上調至臨床效果或最大建議劑量的25-50%。根據處方信息,使用最低有效劑量以達到個別患者治療目標的最短持續時間。在達到穩定狀態(Steady state)之前(對於celecoxib至少8天,對於ibuprofen, flurbiprofen和lornoxicam在PMs中首次劑量後至少5天)不應進行劑量上調。在治療過程中仔細監測不良事件,如血壓和腎功能。或者,考慮使用不由CYP2C9代謝或在體內不受CYP2C9基因變異顯著影響的替代療法。 | 中等建議 | 非主要由CYP2C9代謝的替代療法包括aspirin, ketorolac, naproxen和sulindac。療法的選擇將取決於個別患者的治療目標和毒性風險。 |
| 無法判定 | 不適用c | 攜帶等位基因組合的個體,這些組合不確定和/或功能未知 等位基因的 | *1/*7, *1/*10, *7/*10, *1/*57 | 不適用 | 無建議。 | 不適用 |
a 參見CYP2C9頻率表,了解種族特定的等位基因和表現型頻率。
b 有關CYP2C9 雙倍型和結果表現型的完整列表,請參見CYP2C9基因型至表現型表。
c CPIC為每個等位基因功能狀態分配了一個從0到1的活性值(例如,功能缺失為0,減少為0.5,功能正常為1.0),這些值相加以計算每個雙倍型的活性分數(AS)。CYP2C9 AS已被轉換為表現型分類系統如下:AS為0或0.5的個體為弱代謝型(PMs),得分為1或1.5的為中間代謝型(IMs),得分為2的為正常代謝型(NMs)。
d 評分方案描述於補充資料中。
Celecoxib 途徑, 藥物動力學
概括
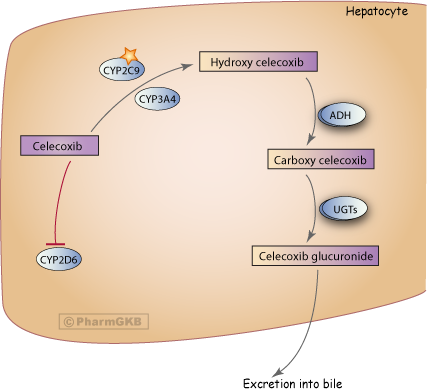
人類肝細胞中Celecoxib的代謝示意圖。
描述
Celecoxib 是一種非類固醇抗炎藥物 (NSAID),具有抗炎、鎮痛和退燒的特性。它已獲准用於治療骨關節炎、類風濕性關節炎、強直性脊椎炎和急性疼痛 (PMID: 10193998; 10804043)。Celecoxib 在癌症預防方面也顯示出潛力,並已用作手術的輔助療法,以減少遺傳性結腸癌易感症候群(家族性腺瘤性息肉病)患者的腺瘤性結腸直腸息肉數量 (FAP) (PMID: 10874062; 16943401; 16943400)。
藥物動力學
經口給藥後,Celecoxib 迅速被吸收,約在 3 小時內達到血清濃度峰值。它在肝臟中被廣泛代謝,幾乎沒有藥物(<3%)以未改變的形式排出體外 (PMID: 10681375)。Celecoxib 的主要排泄途徑為糞便和尿液 (PMID: 18378608)。Celecoxib 主要通過甲基羥基化代謝形成羥基Celecoxib。這一反應主要由 CYP2C9 催化,雖然 CYP3A4 也在其中扮演次要角色(<25%) (PMID: 10749518; 10681375; 12392591)(見圖 1)。羥基Celecoxib 進一步被氧化形成羧基Celecoxib,這一過程通過細胞質醇脫氫酶 ADH1 和 ADH2 進行 (PMID: 12392591),然後通過 UDP 葡萄糖醛酸轉移酶(UGTs)與葡萄糖醛酸結合形成 1-O-葡萄糖醛酸酯。所有代謝物均無藥理活性 (PMID: 10749518)。
由於 Celecoxib 的代謝主要是通過 CYP2C9 媒介,因此 CYP2C9 的多態性可能會直接影響 Celecoxib 藥物動力學 和藥物反應的變異性。那些是 弱代謝型(Poor metabolizer) CYP2C9 底物(例如 CYP2C9*3 等位基因 攜帶者)的人,與正常 CYP2C9 活性的人相比,對 Celecoxib 的暴露增加 (PMID: 11337938; 12392591; 12893985)(見 藥物基因體學 部分)。因此,應謹慎使用抑制 CYP2C9 的藥物於服用 Celecoxib 的患者身上。
雖然不是 CYP2D6 的底物,但 Celecoxib 抑制此代謝酶 (PMID: 12891223)。對於接受 Celecoxib 的患者,應謹慎使用由 CYP2D6 代謝的藥物(例如美托洛爾,(PMID: 12891223)),因為可能存在藥物相互作用的風險。
Celecoxib 途徑, 藥效學
概括
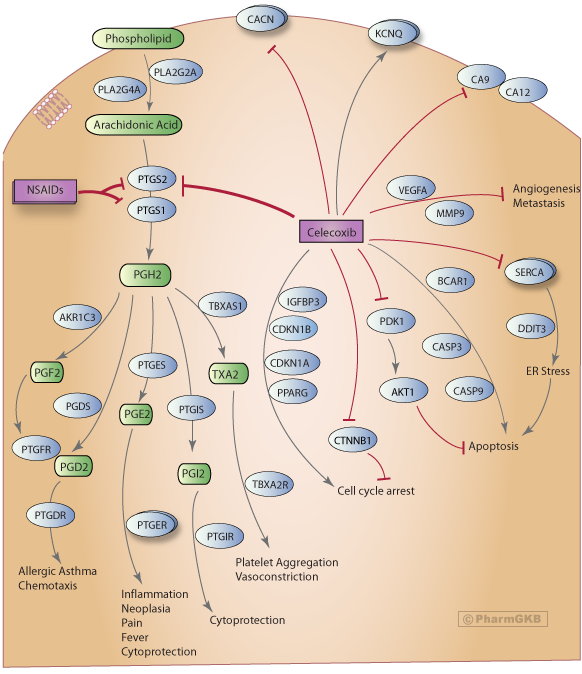
風格化細胞描繪了 Celecoxib 的作用機制,以及與 Celecoxib 互動的候選基因,這些基因參與了 Celecoxib 對細胞週期、凋亡和血管生成的調控。
描述
Celecoxib(Celebrex)是一種選擇性環氧合酶-2(PTGS2/COX-2)抑制劑,用於治療骨關節炎和類風濕性關節炎。它的作用是通過抑制COX-2來減少前列腺素的合成。選擇性擇期 COX-2抑制劑似乎提供與傳統非類固醇抗炎藥物(NSAIDs)相當的抗炎效果,同時避免嚴重的不良反應,特別是由於COX-1(PTGS1)抑制而在長期使用NSAIDs時觀察到的胃腸道毒性。
PK: 口服給藥後,Celecoxib迅速被吸收,約在3小時內達到血清峰值濃度。它在肝臟中被廣泛代謝,只有極少量的藥物(<3%)以未改變的形式排泄(PMID: 10681375)。Celecoxib的主要排泄途徑為糞便和尿液(PMID: 18378608)。Celecoxib主要通過甲基羥基化代謝形成羥基Celecoxib。這一反應主要由CYP2C9催化,雖然CYP3A4也扮演次要角色(<25%)(PMID: 10749518; 10681375; 12392591)(圖1)。羥基Celecoxib進一步被氧化形成羧基Celecoxib,這一過程通過細胞質醇脫氫酶ADH1和ADH2進行(PMID: 12392591),然後通過UDP葡萄糖醛酸轉移酶(UGTs)與葡萄糖醛酸結合形成1-O-葡萄糖醛酸酯。所有代謝物均無藥理活性(PMID: 10749518)。
由於Celecoxib的代謝主要由CYP2C9介導,因此CYP2C9的多態性可能會直接影響Celecoxib 藥物動力學及藥物反應的變異性。與正常CYP2C9活性相比,CYP2C9底物的弱代謝型(Poor metabolizer)攜帶者(例如CYP2C9*3 等位基因)對Celecoxib的暴露增加(PMID: 11337938; 12392591; 12893985)(見藥物基因體學部分)。因此,應謹慎使用抑制CYP2C9的藥物於服用Celecoxib的患者。
雖然CYP2D6的底物,但Celecoxib抑制此代謝酶(PMID: 12891223)。對於由CYP2D6代謝的藥物(例如美托洛爾,(PMID: 12891223)),在接受Celecoxib的患者中也應謹慎使用,以避免潛在的藥物相互作用風險。
PD: Celecoxib通過抑制COX2(PTGS2)來抑制前列腺素的合成。Cox酶(PTGS1和PTGS2)催化從花生四烯酸生成前列腺素的關鍵步驟(PGH2)。PGH2s隨後轉化為活性代謝物(前列腺素E2(PGE2)、前列腺素I2(PGI2)、血栓素(TXA2)、前列腺素D2(PGD2)、前列腺素F2(PGF2)),這些代謝物介導各種生理反應,如炎症、發熱、血壓調節和凝血。PTGS1/COX-1在許多細胞類型中持續表達,而PTGS2/COX-2的表達微不足道,但可以在許多組織中被生長因子、細胞因子和壓力誘導。在如關節炎和癌細胞等炎症性疾病中,PTGS2的水平增加。大多數NSAIDs同時抑制PTGS1和PTGS2,選擇性PTGS2/COX-2抑制劑如Celecoxib(Celebrex)和羅非昔布(Vioxx)已被開發用於治療炎症並提供疼痛緩解。Celecoxib是唯一被批准用於治療家族性腺瘤性息肉病(FAP)的NSAID,這是一種常導致結直腸癌的遺傳性疾病。目前正在探索Celecoxib作為可能的癌症治療。其抗癌活性的確切機制尚不清楚,但最有可能涉及COX-依賴性和COX-獨立的機制。Celecoxib的抗癌機制通常涉及凋亡誘導、細胞週期停滯和血管生成調節。觀察到由Celecoxib介導的細胞週期進程抑制,並伴隨著細胞週期抑制因子CDKN1A/p21和CDKN1B/p27的表達增加,及/或細胞週期蛋白如CCNA1、CCNB1和CCND1的表達減少。在Celecoxib處理的人類結腸癌細胞中也觀察到CTNNB1(β-連環蛋白,促進細胞增殖)的廣泛降解。Celecoxib誘導的凋亡與促凋亡分子的激活(如CASP3、CASP9和DDIT3)和/或抗凋亡分子的抑制(如PDK1及其下游靶標AKT1)相關。Celecoxib的治療還導致VEGFA的表達減少及MMP9的抑制,這表明可能的機制是抑制血管生成和減少腫瘤生長。