CPIC 指南註解:pravastatin 與 SLCO1B1 基因
摘要
對於具有SLCO1B1 功能極低 表現型的患者,建議以≤40mg作為起始劑量,並根據疾病別指南調整pravastatin的劑量。對於SLCO1B1功能降低或可能降低的患者,建議根據疾病別指南開立所需的起始劑量並調整pravastatin的劑量。開藥者應注意pravastatin尤其是在每日劑量超過40mg時,可能增加肌肉病變的風險。
為特定註釋指定基因型或表現型
註釋
此註釋基於CPIC® 指南,針對SLCO1B1、ABCG2和CYP2C9與他汀類藥物相關的肌肉骨骼症狀。
2025年9月
- 應CPIC成員的詢問,SLCO1B1藥物基因專家小組(PCEP)召集會議,重新評估在代表性不足的人群中頻率較高且有新證據顯示與他汀類藥物引起的肌毒性相關的選定等位基因的功能分配。*9和*31的證據摘要和證據強度分配已更新,而*39、*41和*45的功能分配已修訂,以反映其對SLCO1B1轉運蛋白活性影響的新數據。更多更新詳情,請參閱SLCO1B1 等位基因功能參考文件的變更記錄。
2022年2月
-
CPIC指南,針對SLCO1B1、ABCG2和CYP2C9與他汀類藥物相關的肌肉骨骼症狀,已發表於《Clinical Pharmacology and Therapeutics》期刊。CPIC作者總結了文獻,支持如何應用SLCO1B1、ABCG2和CYP2C9基因型測試結果來優化新的或現有的他汀類藥物治療,以降低他汀類藥物相關的肌肉骨骼症狀(SAMS)的風險。當前文件取代了2012年原始指南和2014年針對SLCO1B1和辛伐他汀的更新。此指南新增了針對CYP2C9和ABCG2的建議,並新增了針對所有他汀類藥物的建議。
-
此指南適用於:
- 成人患者
- 兒科患者
-
2022年他汀類藥物劑量指南摘錄:
-
「SLCO1B1促進他汀類藥物以及其他外源性和內源性化合物(例如bilirubin和17-beta-glucuronosyl estradiol)的肝臟攝取。此轉運蛋白的功能減弱(通過基因變異遺傳或藥物介導的抑制獲得)可顯著增加他汀類藥物的全身暴露,這是與SAMS相關的推定因果因素。SLCO1B1基因位於12號染色體(Chr 12p12.2)上,儘管在此基因中已鑑定出許多單核苷酸變異(SNVs),但只有少數已知具有臨床相關的功能影響。(SLCO1B1 等位基因定義和功能表)」
-
「SLCO1B1中最常見且研究最多的變異是c.521T>C(rs4149056),可以單獨基因分型(例如,PCR-based single SNV assay)也可以和其他基因一起,透過多種陣列型檢測平台同時分析。所有SLCO1B1基因測試應檢測c.521T>C;然而,儘管此基因中的其他較不常見的變異可能缺乏行動指導的證據,但它們也可能很重要。」
-
-
下載並閱讀:
表1:基於SLCO1B1 表現型的Pravastatin推薦劑量
改編自2022年指南更新手稿的表1和表2。
| 表現型 | 基因型 | 雙倍型的例子a | 對Pravastatin的影響 | Pravastatin的劑量建議b,c | 建議等級 d |
|---|---|---|---|---|---|
| 功能正常 | 攜帶兩個功能正常 等位基因或一個正常加上一個功能增強 等位基因的個體 | *1/*1, *1/*14 | 典型肌肉病變風險和他汀類藥物暴露 | 根據疾病別指南開立所需的起始劑量並調整劑量。 | 強烈建議 |
| 功能增強 | 攜帶兩個功能增強 等位基因的個體 | *14/*14 | 典型肌肉病變風險和他汀類藥物暴露 | 根據疾病別指南開立所需的起始劑量並調整劑量。 | 強烈建議 |
| 功能減弱 | 攜帶一個正常或功能增強 等位基因加上一個功能缺失 等位基因的個體 | *1/*5, *1/*15 | 與功能正常相比,Pravastatin暴露增加;典型肌肉病變風險,劑量≤40 mg。 | 根據疾病別指南開立所需的起始劑量並調整Pravastatin劑量。開處方者應注意Pravastatin可能增加的肌肉病變風險,尤其是劑量>40mg每天。 | 中等建議 |
| 可能為功能減弱 | 攜帶一個功能缺失 等位基因加上一個不確定/功能未知 等位基因的個體 | *5/*6, *15/*10, *5/*43 | 與功能正常相比,Pravastatin暴露增加;典型肌肉病變風險,劑量≤40 mg。 | 根據疾病別指南開立所需的起始劑量並調整Pravastatin劑量。開處方者應注意Pravastatin可能增加的肌肉病變風險,尤其是劑量>40mg每天。 | 中等建議 |
| 功能極低 | 攜帶兩個功能缺失 等位基因的個體 | *5/*5, *5/*15, *15/*15 | 與正常和功能減弱相比,Pravastatin他汀類藥物暴露增加;典型肌肉病變風險,劑量≤40 mg。 | 開立≤40mg作為起始劑量並根據疾病別指南調整Pravastatin劑量。如果患者能耐受40mg劑量但需要更高效能,可以考慮更高劑量(>40mg)或替代他汀類藥物(參見圖1中替代他汀類藥物的建議)或聯合療法(即Pravastatin加上non-statin類藥物指南指導的醫療療法)。開處方者應注意可能增加的肌肉病變風險,尤其是Pravastatin劑量>40mg。 | 中等建議 |
| 無法判定 | 攜帶一個功能正常 等位基因加上一個不確定或功能未知 等位基因或等位基因與不確定和/或功能未知 等位基因的組合的個體 | *1/*7, *1/*10, *7/*10 | 不適用 | 無建議。 | 無建議。 |
圖1:SLCO1B1建議,根據SLCO1B1 表現型分層的強度和他汀類藥物劑量;所有劑量假設為成人劑量。
改編自2022年指南手稿的圖1
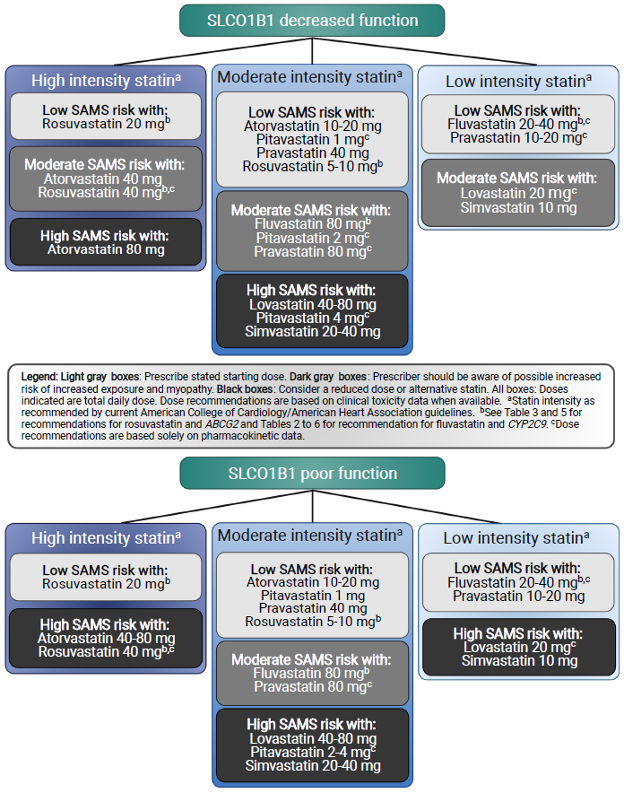
- 「治療建議:SLCO1B1。美國心臟病學會和美國心臟協會於2018年發布了更新的血膽固醇管理臨床實踐指南。在這些指南中,根據預期的LDL-膽固醇降低範圍,將不同每日劑量的他汀類藥物分類為高、中或低強度他汀類藥物。例如,他們建議在有臨床動脈粥樣硬化性心血管疾病(ASCVD)證據的患者中啟動高強度他汀類藥物,這可能包括每天一次40或80 mg的atorvastatin或每天一次20或40 mg的rosuvastatin。圖1設計用於與上述指南結合使用,因為它提供了他汀類藥物建議,包括首選他汀類藥物強度和他汀類藥物劑量,根據SLCO1B1 表現型(即減少或功能極低)分層。淺灰色框中指示的他汀類藥物和劑量可以以最低的SAMS風險開立。深灰色框中指示的他汀類藥物和劑量應謹慎使用(可能增加SAMS風險),而黑色框中指示的他汀類藥物和劑量應避免,因為現有證據表明它們與增加的危害風險相關。這些建議主要基於可用的藥代動力學和SAMS風險數據,並由每種強度內可用的他汀類藥物選擇數量提供信息。」
Ibuprofen 途徑, 藥物動力學
概括
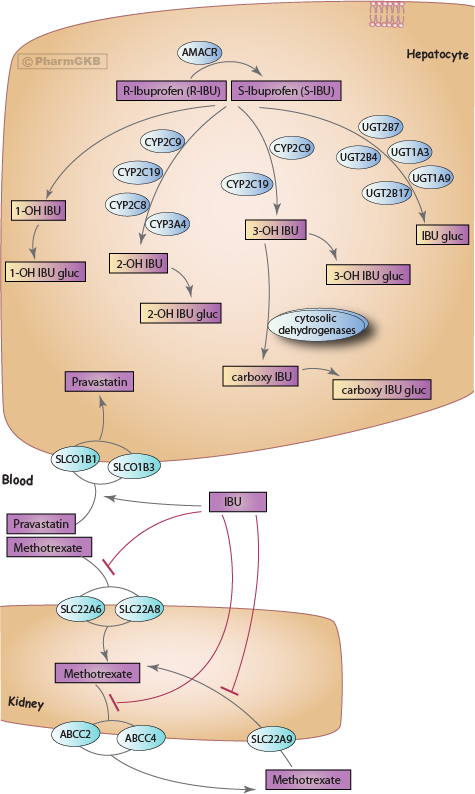
肝臟和腎臟中Ibuprofen的代謝與運輸的風格化圖示。
描述
背景
Ibuprofen (IBU) 是一種傳統的非類固醇抗炎藥物 (NSAID),廣泛用於治療輕度至中等建議的疼痛和炎症。這可能是作為短期的非處方治療,用於頭痛、肌肉疼痛或退燒,或作為長期的TEN處方使用,適用於關節炎和其他慢性病。IBU 抑制由 PTGS1 和 PTGS2 編碼的環氧合酶酶 COX1 和 COX2,防止各種前列腺素的形成(詳情請參見 Celecoxib 途徑 的相關內容)[Article:9515184]。
該藥物以 R 和 S 兩種對映異構體的外消旋混合物形式給藥 [Article:9515184]。口服給藥的 IBU 迅速且完全被吸收,血漿藥物濃度與常用劑量(200-400mg)之間顯示線性關係 [Articles:2109643,9515184]。Ibuprofen 在治療濃度下幾乎完全(>98%)與血漿白蛋白結合 [Article:9515184]。
代謝
IBU 的主要代謝途徑為氧化,涉及細胞色素 P450 酶 [Article:22226725](見圖)。尿液中發現的主要初級代謝物為羧基 IBU 和羥基代謝物 2-OH IBU、3-OH IBU;1-OH IBU 為次要產物 [Article:22226725]。IBU 的羥基和羧基代謝物似乎沒有顯著的藥理活性 [Article:9515184]。IBU 幾乎完全代謝,尿液中幾乎未檢測到未變化的藥物 [Article:22226725]。不同對映異構體的代謝途徑存在差異;S-IBU 的代謝主要通過 CYP2C9 進行,而 R-IBU 則主要通過 CYP2C8 進行 [Article:9296349](見下文討論與不同對映異構體相關的 PGx 影響)。多種其他 CYP 在高濃度的 IBU 下也能進行代謝:CYP3A4、CYP2C8、CYP2C19、CYP2D6、CYP2E1 和 CYP2B6 進行 2-羥基化,CYP2C19 進行 3-羥基化 [Article:18787056]。
估計有 50-65% 的 R-IBU 會轉化為 S 對映異構體 9515184。R-IBU 轉化為 S-IBU 是通過酶 Alpha-methylacyl-coenzyme A racemase 的酰基 CoA 硫酯進行的 AMACR [Article:21614403]。這可以在腸道的系統前進行 [Article:9515184],也可以在肝臟中進行 [Articles:14506641,23376124]。
IBU 的次級代謝通過葡萄糖醛酸化進行,涉及 UGTs [Article:15047194]。體外實驗顯示,重組的 UGT2B7 在與外消旋 IBU 的反應中具有最高活性,但 UGT1A3 和 UGT1A9 也顯示出活性 [Article:15047194]。IBU- 葡萄糖醛酸酯與血漿蛋白的共價結合可能增加毒性的風險 [Article:7781263]。IBU- 葡萄糖醛酸酯在老年人中可佔血漿藥物的 4%,因為清除率降低 [Article:7781263],並且在腎功能不全的個體中也可能升高,增加不良反應的風險 [Article:7714818]。也有報導顯示與硫醇的結合,儘管這些僅佔所有硫醇代謝物的極小部分(所有硫醇合併的尿液代謝物少於 1%)[Article:23052971]。這些代謝物也被認為是反應性的,可能對不良反應有所貢獻,儘管迄今為止的研究主要是在體外或模型生物中進行 [Article:20946099]
運輸
各類運輸蛋白與 NSAID 互動:腎臟和腸胃道中的有機陰離子運輸蛋白(hOAT 家族),以及肝臟有機陰離子運輸蛋白(hOATP 家族)、多藥耐藥蛋白家族的運輸蛋白 (MRPs) 和腸道肽運輸蛋白 (SLC15A1)。 IBU 是一種弱酸且脂溶性,因此它可能能夠在不需要特定運輸蛋白的情況下穿越細胞膜 [Article:9515184]。目前尚不清楚是否有運輸蛋白促進 IBU 的攝取,以及這是否對分佈或清除有影響,但 IBU 與各種運輸蛋白的相互作用對於 藥物交互作用(DDIs)具有臨床重要性 [Article:21389119]。
有機陰離子運輸蛋白 SLC22A6 (hOAT1)、SLC22A7 (hOAT2)、SLC22A8 (hOAT3) 和 SLC22A9 (hOAT4) 能夠在體外攝取 IBU [Article:12388633]。IBU 也與 SLC22A6 (hOAT1) 和 SLC22A8 (hOAT3) 互動,以抑制 methotrexate 的攝取,在 Xenopus 卵母細胞表達系統中進行 [Article:22072415]。對於 SLC22A6,S-IBU 是一種 強烈建議er 抑制劑,而 R-IBU 則不然,但對於 SLC22A8,兩種對映異構體的抑制作用相同 [Article:22072415]。這可能代表了 IBU 和 methotrexate 之間致命 藥物交互作用 的機制,因為 methotrexate 的清除受到抑制,導致毒性血漿藥物濃度 [Article:22072415]。另一種可能的 DDIs 機制是通過 MRP 運輸蛋白 [Article:17005917]。IBU 抑制 ABCC2 (MRP2) 和 ABCC4 (MRP4) 對 methotrexate 的攝取 [Article:17005917]。
雖然 IBU 不是 OATPs 的底物,但 SLCO1B1 (hOATP1B1) 和 SLCO1B3 (hOATP1B3) 仍然與這些運輸蛋白互動 [Article:21389119]。IBU 刺激 pravastatin 的攝取增加,並抑制體外對溴磺酞的攝取 [Article:21389119]。
IBU 也是 SLC15A1 (hPEPT1,未顯示) 在腸道上皮細胞中的非競爭性抑制劑 [Article:20726987]。SLC15A1 運輸蛋白促進各種抗生素和抗病毒藥物以及在光動力癌症治療中使用的伽馬-氨基乙酰酸的攝取 [Article:20726987]。尚不清楚 IBU 是否會在體內與這些藥物產生臨床相關的 DDIs。
不良事件
由於 NSAID 是最常用的藥物類別之一;估計美國有 6-24% 的成年人在某個月份使用過 NSAID,因此安全性至關重要 [Article:20101062]。對於 IBU,臨床上最關注的不良事件是胃腸道(GI)出血或潰瘍,以及心血管(CV)事件。關於個別 NSAID 的數據可能難以獲得。雖然在使用 Ibuprofen 的情況下報導了罕見的嚴重皮膚疾病,如 史蒂芬-強森症候群及毒性表皮壞死溶解症(SJS/TEN),但這些情況極為罕見,對於大多數 NSAID 的發生率低於每百萬用戶每週 1 例 [Article:20101062]。
設計 擇期 COX2 抑制劑或 coxib 類藥物的原因之一是傳統 NSAID 的胃腸道風險 [Article:23163547]。然而,幾項大型研究和 綜合分析(Meta-analysis) 表明,短期每日使用 IBU 的胃腸道副作用風險輕微,且與對乙醯氨基酚或安慰劑之間沒有顯著差異 [Article:23163547]。藥物基因組學研究表明,這些風險可能受到基因變異的調節 [Articles:17681167, 18216720, 20445534]。
在 rofecoxib 退市後,TEN 對所有 NSAID 的相對心血管風險引起了關注。一項最近的系統評價研究了三十九項涉及 IBU 的研究,發現低劑量下心血管事件的比值比為 1.18(1.11-1.25),或為「低風險」 [Article:21980265]。然而,在較高劑量下 IBU 確實增加了心血管事件的風險。在成對比較中,IBU 的風險顯著低於雙氟氯噻嗪和依托考昔,且略高於但仍顯著高於萘普生 [Article:21980265]。
藥物基因體學
在表達蛋白的研究中,CYP2C9 偏向於 S-2-OH IBU 和 S-3-OH IBU 的形成,而 CYP2C8 偏向於 R-2-OH IBU 的形成 [Article:9296349]。因此,不同 IBU 代謝物的相對量可能因 CYP2C9 和 CYP2C8 的變異而改變,這是由於這種立體特異性偏好。在一項對 30 名健康白人波蘭志願者的研究中,IBU 的清除率在具有 等位基因 功能缺失的個體中降低 CYP2C9 和 CYP2C8 [Article:19480553]。那些具有 CYP2C9*3 (n=7) 或 CYP2C8*3 (n=5) 的個體,其外消旋混合 IBU 的清除率低於 CYP2C9*1 和 CYP2C8*1 (n=16),而那些同時具有 CYP2C8*3 和 CYP2C9*2 (n=2) 的個體則有顯著較低的清除率(沒有個體具有 CYP2C9*2 而沒有 CYP2C8*3 等位基因)。[Article:19480553]。當單獨考慮 R-IBU 時,CYP2C8*3 的血漿藥物濃度顯著較低。具有 CYP2C8*3 和 CYP2C9*2 的個體也有顯著較低的血漿羧基 IBU,而 2-OH IBU 的量在所有組別中相似 [Article:19480553]。
一些研究表明,這種清除率的降低導致持續的 IBU 水平可能增加胃腸事件的風險。在一項小型意大利 NSAID 使用者的研究中(n=26,其中 3 名為 IBU 使用者),出現胃十二指腸出血的頻率顯著高於對照組的 CYP2C9*1/*2 和 CYP2C9*1/*3 基因型 [Article:17681167]。在一項西班牙的病例對照研究中,CYP2C9*2 與出血風險的關聯也得到了確認(n=134,其中 14 名為 IBU 使用者) [Article:18216720]。在西班牙的研究中,CYP2C8*3 也與 NSAID- 相關的出血風險有關,結合 CYP2C8*3-CYP2C9*2 單倍型 的風險進一步增加 [Article:18216720]。在法國的一項研究中,代表非阿斯匹靈 NSAID 使用者的各種族群(n=57,其中 11 名為 IBU 使用者)未重複 CYP2C8 的關聯,但確實觀察到 CYP2C9*3 與急性上消化道出血的風險增加 [Article:20445534]。
迄今為止,有一篇論文研究了 IBU 的療效和 PGx:檢查基因變異和在智慧牙拔除手術後使用 IBU 或 rofecoxib 的疼痛感知 [Article:16678543。在手術後 48 小時,具有 rs20417] 的 GG 或 CG 基因型的患者在使用 IBU 時顯示出顯著的疼痛減輕,相較於 rofecoxib,而具有 CC 基因型的患者對 rofecoxib 的反應優於 IBU [Article:16678543(注意:該基因位於染色體的負鏈上,在 ClinPGx 中補充至正鏈,在該論文中報導為負鏈)。rs20417 變異位於啟動子區域,影響轉錄因子的結合。在具有 rs20417] CC 基因型的個體中,PTGS2,因此 COX2-s擇期 抑制劑可能對疼痛的影響大於具有 GG 或 CG 基因型的個體,後者的 PTGS1 和傳統 NSAID 如 IBU 更為有效。
有關 IBU 的一個有趣的 PGx 領域是該藥物如何與 AMACR 變異體互動並調節癌症風險 [Article:23376124]。到目前為止,尚無研究將 IBU 與 AMACR 的相互作用與 AMACR 的基因變異聯繫起來。AMACR 是負責將 R 轉化為 S 的酶 IBU。AMACR 也與棕櫚酸相互作用,棕櫚酸是一種在紅肉和乳製品中發現的膳食成分,可能在其解毒中發揮作用。AMACR 蛋白質在前列腺 癌細胞 和其他多種癌症中的水平增加。AMACR 的變異和替代剪接形式與癌症相關。目前尚未顯示有變異與 IBU 之間存在不同的相互作用。
pravastatin 途徑, 藥物動力學
概括
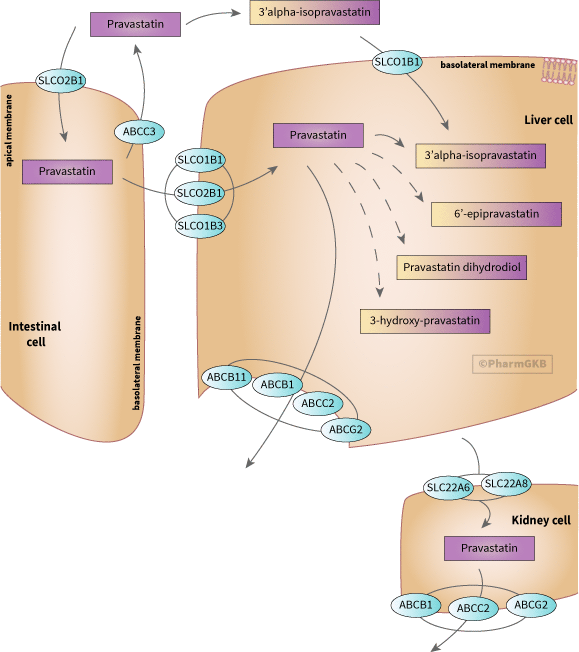
涉及pravastatin的運輸和代謝的候選基因表示。
描述
背景
pravastatin是一種HMG-CoA還原酶抑制劑或他汀類藥物,用於預防高膽固醇血症患者的心血管疾病。與其他他汀類藥物類似,pravastatin通過對3-羥基-3-甲基戊二酸輔酶A還原酶(HMGCR)的競爭性抑制來抑制內源性膽固醇的生成,該酶催化HMG-CoA轉化為美伐酸,如他汀途徑、藥效學所示。
有關使用藥物基因組學信息進行pravastatin劑量調整的指南由臨床藥物基因體學實施聯盟(CPIC)發布。CPIC建議對於SLCO1B1代謝不良或功能減弱表現型的患者減少pravastatin的起始劑量。
代謝
pravastatin以活性形式給藥,並可在肝臟中廣泛代謝為活性和非活性代謝物[Article:10490896]。然而,pravastatin是與羅蘇伐他汀和匹伐他汀一起的親水性他汀類藥物,主要以未改變的形式排泄[Article:17178259]。
運輸
pravastatin是有機陰離子轉運蛋白SLCO1B1、SLCO1B3和SLCO2B1的底物[Articles:16198652、15970799、15864131、16103896、15564882、15226675],以及SLC22A6和SLC22A8 [Article:12811365]。它也是外排轉運蛋白的底物,如ABCB1、ABCB11、ABCC2、ABCC3和ABCG2 [Articles:34162690、16107564、15901800、15901796、15616150、15226675、11902809]。
在SLCO1B1基因中的遺傳變異與pravastatin的暴露增加和清除減少有關。攜帶rs4149056 (521T>C) C 等位基因的患者,其AUC的pravastatin增加[Articles:12811365、15116054、15226675、16678544、17047488、17622941、18408565、19776292、30549267、26744986、17622941],這可能也會導致他汀類藥物相關肌肉症狀的風險增加。然而,在STRENGTH(他汀反應由基因單倍型標記檢查)研究中,rs4149056與接受pravastatin的個體的不良事件並無顯著關聯[Article:19833260]。rs4149056對pravastatin的療效影響也微乎其微[Articles:22189199、16678544、17439540、25379722]。