CPIC 指南註解:aminosalicylic acid、chloramphenicol、chloroquine、ciprofloxacin、dimercaprol、doxorubicin、furazolidone、glyburide、hydroxychloroquine、mafenide、nalidixic acid、norfloxacin、ofloxacin、phenazopyridine、quinine、sulfadiazine、sulfamethazine、sulfamethoxazole、sulfamethoxazole / trimethoprim、sulfanilamide、sulfasalazine、sulfisoxazole、tolbutamide、vitamin c、Vitamin K and analogues 與 G6PD 基因
摘要
對於具有G6PD 缺乏或G6PD 可變型(Variable) 表現型的患者,沒有理由避免使用低至無風險的藥物。在患有慢性非球形紅細胞溶血性貧血(chronic non-spherocytic hemolytic anemia, CNSHA)的G6PD 缺乏患者中,應謹慎使用低至無風險的藥物,並建議密切監測慢性溶血的急性加重。然而,CPIC不建議基於G6PD基因型改變低至無風險藥物的處方行為。
註釋
此註釋基於擴展的CPIC®指引,針對G6PD基因型的藥物使用。CPIC作者評估了攜帶G6PD變異的患者使用各種藥物的現有證據。
2022年9月
-
擴展的CPIC®指引,針對G6PD基因型的藥物使用已在《Clinical Pharmacology and Therapeutics》期刊中發表。作為該指引的一部分,作者將藥物按其在G6PD缺乏症患者中引起急性溶血性貧血(AHA)的風險進行分類。4-aminosalicylic acid, chloramphenicol, chloroquine, ciprofloxacin, dimercaprol, doxorubicin, furazolidone, glyburide, hydroxychloroquine, mafenide, nalidixic acid, norfloxacin, ofloxacin, phenazopyridine, quinine, sulfadiazine, sulfadimidine, sulfamethoxazole, sulfanilamide, sulfasalazine, sulfisoxazole, tolbutamide, vitamin C和vitamin K被分類為低至無風險藥物,如指引發表的表2所示。CPIC不建議基於基因型改變低至無風險藥物的處方行為,並且G6PD/低至無風險藥物基因/藥物對被CPIC分配為C級最終狀態。
-
這些指引適用於:
- 成人患者
- 兒科患者
-
指引摘錄:
- 「在此CPIC指引中增加了一個步驟,將藥物分為三組:在G6PD缺乏症存在下可被視為高風險的藥物(因此通常應避免),在G6PD缺乏症中被視為中等風險的藥物(因此應謹慎使用),以及可被視為低至無風險的藥物(在G6PD缺乏症患者中不會增加AHA風險)。」
- 「為了將藥物分配到風險組,作者不僅考慮了主要同行評審文獻中的證據強度,還考慮了藥物使用頻率、監管機構警告的存在以及是否存在可能產生活性氧物質並在G6PD缺乏症中促成溶血的機制(補充,分配風險等級)」
- 「Sulfamethoxazole(低至無風險藥物) 在G6PD缺乏症中sulfamethoxazole的風險類別。 Sulfamethoxazole被認為屬於低至無風險類別,因為將其與AHA聯繫起來的證據較弱(主要是病例報告,常常因感染、蠶豆或其他藥物的存在而混淆),且有幾項研究顯示在G6PD缺乏症中安全使用。一項研究報告顯示在10名缺乏嬰兒中未見此藥引起AHA。儘管有一些監管機構的警告,但大多數較弱。此外,sulfamethoxazole被廣泛使用,進一步證明缺乏支持顯著風險的研究。」
-
下載並閱讀:
- 擴展的藥物基因體學臨床應用聯盟(CPIC)指引,針對G6PD基因型的藥物使用
- 2022補充
- G6PD 基因資訊表
- 藥物資源映射
- 4-aminosalicylic acid
- chloramphenicol
- chloroquine
- ciprofloxacin
- dimercaprol
- doxorubicin
- furazolidone
- glyburide
- hydroxychloroquine
- mafenide
- nalidixic acid
- norfloxacin
- ofloxacin
- phenazopyridine
- quinine
- sulfadiazine
- sulfadimidine
- sulfamethoxazole/trimethoprim
- sulfanilamide
- sulfasalazine
- sulfisoxazole
- tolbutamide
- vitamin C
- vitamin K
- 測試前後警示
- 4-aminosalicylic acid
- chloramphenicol
- chloroquine
- ciprofloxacin
- dimercaprol
- doxorubicin
- furazolidone
- glyburide
- hydroxychloroquine
- mafenide
- nalidixic acid
- norfloxacin
- ofloxacin
- phenazopyridine
- quinine
- sulfadiazine
- sulfadimidine
- sulfamethoxazole/trimethoprim
- sulfanilamide
- sulfasalazine
- sulfisoxazole
- tolbutamide
- vitamin C
- vitamin K
- 低至無風險藥物流程圖
表1:低至無風險藥物在G6PD 表現型中的推薦治療使用
改編自指引的表1、2和6
| 預測表現型 | 基因型a | 基因型範例b | 影響 | 治療建議 | 建議等級c | 考量 |
|---|---|---|---|---|---|---|
| 正常 | 攜帶一個X染色體,帶有非缺乏(IV類)等位基因 或攜帶兩個非缺乏(IV類)等位基因者 |
B, Sao Boria, IV B/B, B/Sao Boria, B/A, IV/IV |
急性溶血性貧血的低至無風險 | 基於G6PD狀態,無理由避免低至無風險藥物 | 強烈建議 | |
| 缺乏 | 攜帶一個X染色體,帶有缺乏(II-III類)等位基因 或攜帶兩個缺乏(II-III類)等位基因者或一個I類等位基因和一個II或III類等位基因者 |
A-, Orissa, Kalyan-Kerala, Mediterranean, Canton, Chatham, II, III A-/A-, A-/Orissa, Orissa/ Kalyan-Kerala, Mediterranean/ Mediterranean, Chatham /Mediterranean, Canton/ Viangchan, II/II, II/III, III/III, I/II, I/III |
急性溶血性貧血的低至無風險 | 基於G6PD狀態,在標準劑量下無理由避免低至無風險藥物 | chloramphenicol, chloroquine, hydroxychloroquine, vitamin C, vitamin K: 中等建議 所有其他低至無風險藥物: 可選建議 |
對於高於正常劑量的情況,並在感染或其他氧化壓力的情況下,包括同時使用多種中等和低至無風險藥物時,可能需要更密切的監測。 |
| 嚴重缺乏合併慢性非球形紅細胞溶血性貧血(Deficient with CNSHA) | 攜帶一個X染色體,帶有缺乏(I類)等位基因 或攜帶兩個缺乏(I類)等位基因者d |
Bangkok, Villeurbanne, I Bangkok/Bangkok, Bangkok/Villeurbanne, I/I |
慢性溶血性貧血急性加重的高風險 | 在此組中應謹慎使用所有藥物;如果使用藥物,建議密切監測慢性溶血性貧血的急性加重。 | 可選建議 | 在G6PD 嚴重缺乏合併慢性非球形紅細胞溶血性貧血(Deficient with CNSHA) 表現型患者中數據不足,但由於潛在的病理生理學賦予慢性溶血性貧血急性加重的高風險,應仔細權衡使用任何藥物的風險與益處。 |
| 可變型(Variable)e | 攜帶一個非缺乏(IV類)等位基因和一個缺乏(I-III類)等位基因者 | B /Bangkok, B/Mediterranean, B/A-, IV/I, IV/II, IV/III | 急性溶血性貧血的低至無風險 | 基於G6PD狀態,在標準劑量下無理由避免低至無風險藥物 | 中等建議 | 由於X 染色體聯鎖嵌合現象(X-linked mosaicism),攜帶多於一個X染色體的個體(例如,女性,克氏綜合症患者)和異型合子(Heterozygous)一個非缺乏(IV類)和一個缺乏(I–III類)等位基因者可能顯示正常或缺乏 表現型。 |
| 無法判定 | 攜帶至少一個等位基因具有功能尚未確定 | Dagua B/Dagua |
急性溶血性貧血的未知風險 | 為確定G6PD狀態,必須測量酶活性。藥物使用應根據基於活性表現型的建議進行指導。 | 中等建議 |
CNSHA:慢性非球形紅細胞溶血性貧血
a WHO分類來自[文章:22293322], 其他細節來自[文章:4963040]。I類等位基因極為罕見;II類和III類等位基因之間的區分不明顯。幾乎所有患者都攜帶II、III或IV類等位基因。
b 由於G6PD 等位基因的數量龐大,除了此處給出的範例外,還可能存在其他基因型;請參閱G6PD 等位基因定義表以獲取更全面的等位基因和G6PD 等位基因功能表以了解其分配的功能(WHO類別)。請注意,一些實驗室使用“B 等位基因”的標示來指示不攜帶已知I-III類變異的等位基因。可參考G6PD頻率表以了解G6PD 等位基因在主要生物地理群體中的頻率。
c 評分方案在指引補充的建議強度部分中描述。
d 此類基因型從未見過,推測極為罕見。
e 由於X 染色體聯鎖嵌合現象(X-linked mosaicism),攜帶一個非缺乏(IV類)和一個缺乏(I-III 等位基因)等位基因的個體(通常為女性)可能顯示正常或缺乏 表現型。因此,很難預測這些個體的表現型(見補充,G6PD異合子)。
Clozapine 途徑, 藥物動力學
概括
肝臟中 clozapine 代謝與運輸之示意圖
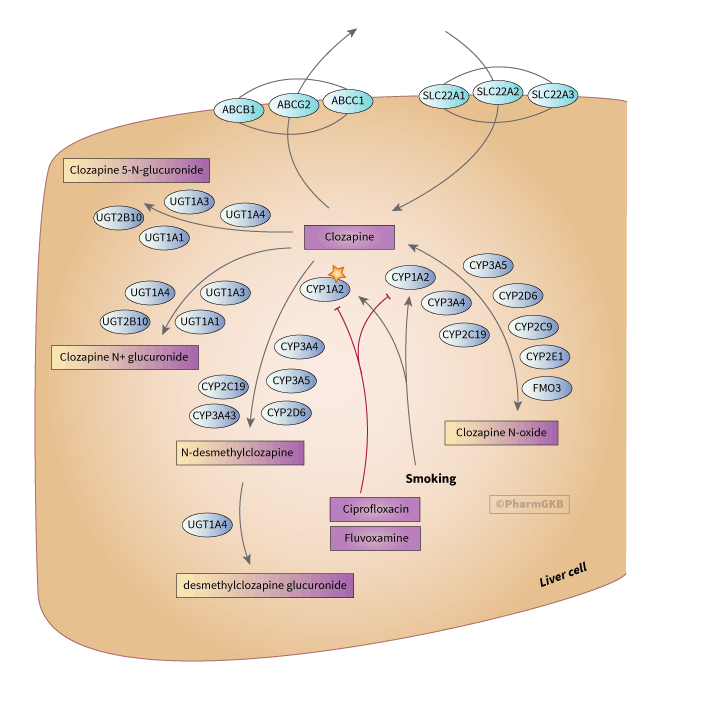
描述
背景
Clozapine是一種非典型抗精神病藥物,也是治療難治性精神分裂症的金標準(TRS),即至少有兩種傳統抗精神病藥物無效的患者[Article:27932669]。指導方針僅允許在TRS中開立Clozapine的原因是其罕見但潛在致命的Clozapine誘導的顆粒細胞缺乏症(CIA)[Article:27932669]。FDA 仿單還列出了黑框警告,指出觀察到與劑量相關的副作用,包括直立性低血壓、心動過緩、暈厥和癲癇發作,並警告老年人有嚴重心臟副作用和死亡率增加的風險(請參見藥物仿單)。其他常見副作用包括鎮靜(大多為暫時性)和代謝性副作用,如體重增加。雖然所需劑量在患者之間可能有很大差異(約150 – 1000mg/天),Clozapine的治療範圍較窄,建議進行治療藥物監測(TDM):Clozapine的血清濃度低於250 ng/mL與復發相關,而高於750 ng/mL則與中毒風險增加相關[Article:27932669。目前尚無針對影響Clozapine代謝的變異體的已發表藥物基因組學(PGx)指導方針,FDA 仿單對CYP2D6 弱代謝型(Poor metabolizer)和潛在的藥物交互作用與同樣CYP450酶代謝的藥物的相互作用提出警告。這個摘要檢視了藥物動力學(PK)Clozapine的代謝及其相關候選基因,並討論了其變異的影響。雖然Clozapine作用於大量受體,可能多達39種不同的受體PDSP Ki數據庫],但藥效學(PD)Clozapine的影響不在本文範疇內,除非PD效應是PK基因的結果。
代謝
Clozapine經過廣泛的肝臟代謝,主要途徑為去甲基化至n-desmethylclozapine和氧化至clozapine n-oxide(如上圖所示)[Article:7891353。體外實驗表明,CYP3A4約占Clozapine清除的70%,CYP1A2約占15%,而CYP2C19、CYP2C8和[[KEEP_358FDA2f]]各占5%或更少[Article:27320963]。對Clozapine代謝的體外研究表明,CYP3A4和CYP1A2是主要負責去甲基化的酶,而CYP2D6的作用非常小[Article:7891353][Article:18809730]。幾種酶能夠在體外生成Clozapine的n-氧化物代謝物(CYP1A2、CYP2E1、CYP2C9、CYP3A4、CYP2D6、FMO3、CYP2C19),但在體內CYP1A2被認為是主要催化劑[Article:7891353][Article:18809730][Article:27932669][Article:23297297]。體內研究表明CYP3A5和CYP3A43也可能發揮作用[Article:28340122][Article:25150845]。n-desmethylclozapine是一種活性代謝物,能夠通過多巴胺D2和D3受體發揮作用[Article:19483482],組胺受體[Article:21912901],毒蕈鹼M1[Article:25859763],以及血清素受體[Article:17583355]。此外,Clozapine與n-desmethylclozapine的比率是預測精神分裂症患者工作記憶表現的強烈建議指標;較低的比率與更好的工作記憶和執行功能相關[Article:25859763][Article:29024893]。clozapine n-oxide被認為是無活性的,可能會被代謝回Clozapine [Article:7891353][Article:16805946][Article:28664816]。在接受Clozapine治療的患者尿液中已識別出額外的代謝物,但這些代謝物的臨床意義尚不清楚[Article:9833598]。
雖然在CYP1A2中存在功能變異體,但對這些變異體的影響尚無共識,特別是與Clozapine相關[Article:15206669][Article:15949157][Article:16044115][Article:21481946][Article:25090458]。吸煙者的Clozapine血清濃度低於非吸煙者,這是由於CYP1A2的活化[Article:11476124]。吸煙行為的變化可以顯著改變Clozapine的代謝;在戒煙後,Clozapine的暴露增加超過50%[Article:11763003]。強烈建議的CYP1A2抑制劑,如抗生素ciprofloxacin,也可以顯著增加Clozapine的暴露,並且病例研究報告了致命的相互作用[Article:27872784]。此外,在感染期間,促炎細胞因子可以下調CYP1A2的表達,這可能會加劇這一效應[Article:25519488]。抗抑鬱藥fluvoxamine也是CYP1A2的抑制劑,並且可以增加Clozapine的血漿濃度。這一PK效應可以用作調節Clozapine劑量的機制;減少Clozapine的劑量以嘗試減少副作用,同時保持療效[Article:10982203]。
總體而言,劑量、性別、年齡和吸煙約占血清Clozapine變異性的50%,其餘50%可能由藥物代謝酶的基因變異和共同用藥組成[Article:9798077][Article:2813665]。男性的血清Clozapine濃度低於女性,儘管女性接受的劑量較低[Article:2813665][Article:8195452][Article:9798077]。吸煙者的血漿Clozapine濃度約低20-30%相比非吸煙者[Article:2813665][Article:8195452][Article:9798077]。年長患者(45歲以上)的濃度顯著高於年輕患者(18-26歲)[Article:2813665]。一項針對接受抗精神病藥物的孕婦的小型研究發現,雖然對Clozapine(n=4)的血漿濃度下降並不顯著,但對其他藥物(阿立哌唑n=14和奎etiapine n=35)則顯著,作者推測在懷孕期間CYP3A4的水平會增加,這可能會影響抗精神病藥物的PK[Article:28643331]。這與懷孕期間行為的改變(如咖啡因使用和吸煙的變化)相結合,可能對接受Clozapine的女性進行監測非常重要。
運輸
關於Clozapine運輸的實驗涉及多種細胞類型,尚未提供關於哪些運輸蛋白參與進入或排出的統一圖景。以下是分散的證據,所有合理的候選者在圖1中顯示:
Clozapine有效地穿越血腦屏障並進入大腦,如PET對C11的成像所示仿單的Clozapine [Article:26225256]。然而,這項研究使用了靜脈注射Clozapine,而大多數患者是口服給藥,且負責的運輸蛋白未被確定。
對於口服Clozapine,腸道的運輸非常重要。有證據表明,Clozapine在腸道的代謝與在肝臟的代謝不同,因為葡萄柚汁(腸道CYP3A4的抑制劑,但不是肝臟CYP3A4的抑制劑)不影響血漿Clozapine濃度[Article:11791889][Article:11816871]。
肝臟中的攝取可能由SLC22A1催化,因為Clozapine能夠在體外抑制SLC22A1對模型底物(MPP+)的運輸[Article:22806583]。在同樣的體外實驗中,Clozapine也與SLC22A2和SLC22A3相互作用[Article:22806583]。
Clozapine與ABCB1編碼的運輸蛋白(也稱為PgP)之間的關係在文獻中存在矛盾。早期的體外實驗使用CaCo2細胞(腸道細胞系)顯示Clozapine和n-desmethylclozapine是ABCB1的低親和力底物,並且是ABCB1-介導的塔尼洛洛運輸的抑制劑[Article:15285840]。然而,儘管ABCB1變異體與不同的PK或結果相關[Article:19593168][Article:28919802],最近的文章報導Clozapine不是ABCB1的底物或有效抑制劑[Article:21843066]。
Clozapine在體外抑制ABCG2對米托坦的運輸[Article:18834354]。此外,ABCG2運輸蛋白的變異體與Clozapine的暴露相關,而ABCC1的變異體與結果相關,但尚未明確顯示Clozapine是兩者的底物(在PGx部分進一步討論)[Article:27932669][Article:28919802]。
藥物交互作用,DDIs
有關Clozapine 藥物交互作用的出版物很多,但大多數研究規模很小或為個案報告,且很少考慮PGx變異體是否影響相互作用(在[Article:17214606]中回顧)。 FDA 仿單對Clozapine的分類將DDI分為四種類型,並對每種類型提出建議:
- 強烈建議 CYP1A2抑制劑,例如fluvoxamine和ciprofloxacin,建議將Clozapine劑量減少至三分之一。
- 中等建議或弱CYP1A2抑制劑,如口服避孕藥或咖啡因,建議監測不良反應並考慮減少劑量。
- CYP2D6或CYP3A4抑制劑,如氟西汀,建議監測不良反應並考慮減少劑量。
- 強烈建議 CYP3A4誘導劑不建議使用,例如卡馬西平、利福平、苯妥英和聖約翰草。 每種類型的DDI研究在下文中討論並在表1中總結:
類型1 DDIs:強烈建議 CYP1A2抑制劑
文獻中有多篇報告顯示Clozapine與ciprofloxacin之間存在嚴重且有時致命的相互作用,支持調整Clozapine劑量的建議[Article:27872784][Article:23346515][Article:22929408][Article:19067475][Article:18354073][Article:17624521][Article:17329613][Article:11151749]。還有大量文獻關於fluvoxamine和Clozapine(在[Article:17214606]中回顧):幾篇論文討論了使用fluvoxamine作為輔助藥物以避免高劑量Clozapine;較低的Clozapine劑量減少了包括體重增加[Article:25627831]、葡萄糖/代謝功能障礙[Article:28688742]、和血液毒性[Article:23490199]的副作用發生率。 一項使用人類肝臟微粒體的體外研究調查了與fluvoxamine共同治療時n-desmethylclozapine和clozapine n-oxide的產生。作者得出結論,咖啡因表型在預測CYP1A2-相關的Clozapine-fluvoxamine DDI時並不可靠[Article:29155491]。
類型2 DDIs:中等建議或弱CYP1A2抑制劑
咖啡因攝入是血清Clozapine水平的重要預測因子[Article:23104241]。各種案例研究顯示,咖啡因攝入可以影響Clozapine的血漿水平和Clozapine的療效[Article:8601555][Article:17515710]。一項隨機研究交替讓患者飲用常規咖啡或無咖啡因咖啡,顯示咖啡因與血漿Clozapine相關[Article:14725610]。咖啡因攝入也可以來自可樂飲料,其中一天六杯的攝入在一個案例研究中使血漿Clozapine達到毒性水平[Article:22926611]。個體對咖啡因的血漿Clozapine反應的變異受到CYP1A2的影響[Article:14725610]。
有幾個案例報告顯示DDI與Clozapine和口服避孕藥的相互作用[Article:12454563][Article:25890012][Article:24717251]。在現實情況中,監測和考慮劑量調整的建議可能會更複雜,因為開處方的醫生可能不知道患者的避孕藥處方歷史,而患者也可能未意識到分享這些信息的重要性。
類型3 DDIs:CYP2D6或CYP3A4抑制劑
與由CYP2D6代謝的抗抑鬱藥(包括舍曲林、帕羅西汀和氟西汀)共同治療的Clozapine報導導致血漿Clozapine增加20-40%(在[Article:24494611]中回顧)。雖然Centorrino的研究檢查了40名與SSRIs共同治療的患者和40名單獨接受Clozapine的患者,顯示血清Clozapine和n-desmethylclozapine增加,但血清濃度的標準差非常大(例如,與帕羅西汀共同治療的患者的平均Clozapine濃度為417ng/ml,標準差為373ng/ml)[Article:8633698]。另一項對14名接受帕羅西汀和Clozapine的患者的研究顯示Clozapine或主要代謝物沒有變化[Article:9928923][Article:9472836]。在一個個案報告中,一名接受Clozapine和帕羅西汀的患者血清Clozapine出現毒性增加{Joos, 1997 #104}。而一項對9名與帕羅西汀共同治療的患者和8名與舍曲林共同治療的患者的研究發現,僅在帕羅西汀組中Clozapine和N-去甲基Clozapine增加[Article:11147928。然而,由於帕羅西汀也由CYP1A2和CYP3A4代謝,因此這一關係可能並不完全由CYP2D6的抑制引起,請參見帕羅西汀途徑]。 在一項體外微粒體實驗中,CYP2D6的抑制劑奎尼丁和右美沙芬影響Clozapine的代謝,並減少了次要代謝產物的生成,如HPLC所示,但未改變n-desmethylclozapine或clozapine n-oxide的量[Article:1545398]。
類型4 DDIs:強烈建議 CYP3A4誘導劑
雖然不建議共同開處方Clozapine和強烈建議 CYP3A4誘導劑,但仿單列舉了如苯妥英和卡馬西平的例子,但不包括奧卡西平。關於奧卡西平的案例報告表明,它可能特別容易通過Clozapine與CYP3A4和CYP1A2相互作用[Article:28255440][Article:26711482]。
其他DDIs
Clozapine的副作用之一是胃食道逆流病,GERD [Article:28291036]。GERD的治療是氫離子幫浦阻斷劑(PPI)(PPIs),其中大多數由CYP2C19代謝。DDI與Clozapine和PPIs之間有幾種可能的相互作用路徑;CYP1A2的誘導、FMO3的誘導,以及CYP2C19的競爭性抑制。這些相互作用中的任何一種或全部都可能導致n-desmethylclozapine和clozapine n-oxide的增加,並增加血液毒性的風險[Article:28291036]。clozapine n-oxide在體外抑制CYP2C19介導的-美芬妥因的代謝[Article:27853934]。因此,clozapine n-oxide可能會抑制PPIs的代謝。
中等建議或弱CYP1A2或CYP3A4誘導劑也在藥物仿單中討論,建議在觀察到療效不足時增加劑量。
藥物仿單還警告在已經有長QT風險的人群中使用Clozapine,並提到許多可能增加長QT綜合症風險的藥物。列出的幾種藥物也被禁用作CYP3A4的抑制劑,例如紅霉素或CYP2D6的抑制劑,例如奎尼丁、氯丙嗪或CYP1A2的抑制劑,例如胺碘酮。
藥物基因體學
雖然大多數有關Clozapine的PGx研究已檢查PD候選基因,但本文專注於PK候選基因及其對PK或PD/臨床結果的影響(在表2中總結)。候選基因CYP1A2、CYP3A4、CYP2C19、CYP2D6和ABCB1都是知名的藥物基因。此外,還有詳細描述每個PGx關係的論文,這些論文合併到變異註釋標籤下的互動表中。
CYP1A2
目前尚無與CYP1A2代謝相關的編碼序列變異,這可能是由於在大多數人群中的頻率非常低(在{Thorn, 2012 #63}中回顧,並在CYP1A2 VIP摘要中總結)。最常研究的變異位於上游啟動子區域:CYP1A2:(-163)C>A (rs762551),CYP1A2:(-3860)G>A (rs2069514)和CYP1A2:(-729)C>T (rs12720461),這些變異形成了單倍型 *1F、*1C、*1D和*1K [Article:21989077]。由於歷史上對於-164(rs762551)哪個變異代表*1F的混淆,具體變異在報告中給出。單倍型 *1F被認為是一種可誘導的變異,具有高活性,單倍型 *1C、*1D和*1K被認為是低活性[Article:21989077]。
在接受Clozapine的患者中,CYP1A2*1F與治療不反應相關,需要更高劑量[Article:28356835][Article:15206669][Article:16044115][Article:15949157]。CYP1A2 *1F(rs762551 AA)的純合子吸煙者的血漿Clozapine較低,代謝物濃度較高,清除速度較快,與非吸煙者相比。在一項針對4名對Clozapine無反應的重度吸煙者(每天30支或更多香煙)的案例研究中,發現他們都是同型合子(Homozygous) *1F(描述為-164C > A,均為AA)[Article:15206669]。所有患者在Clozapine血漿水平提高至治療閾值以上後,無論是通過將Clozapine劑量提高至非常高的值,還是通過引入fluvoxamine,均經歷了臨床狀態的顯著改善[Article:15206669]。另一個案例報告中,兩名患者戒煙後出現嚴重不良反應:一名吸煙超過40支/天的患者突然戒煙,血漿Clozapine極高且出現不良反應;另一名報告每天吸煙3或4支的患者住院且不被允許吸煙,並有多種共同用藥。兩名患者均為同型合子(Homozygous) CYP1A2*1F,具體定義為-164AA[Article:16044115]。此外,對58名接受Clozapine的精神分裂症患者的研究發現,吸煙者的濃度劑量比(C/D)比CYP1A2*1F(AA)低,但這並不顯著[Article:15949157]。非吸煙者的*1F雜合子或純合子有較高的C/D比率。吸煙者被描述為每天至少吸煙15支,但範圍未報告[Article:15949157][Article:12618594]。
接受Clozapine的患者在劑量較低(< 300 mg/d)時,癲癇風險較低,而在維持劑量較高(>或= 600 mg/d)時,癲癇風險較高[Article:7991106]。同型合子(Homozygous) CYP1A2*1F基因型與接受Clozapine的患者癲癇風險增加相關(n=108)[Article:23601795],這可能反映了CIA*1F與高劑量的關聯。
表達低的等位基因的CYP1A2患者更可能出現與血漿Clozapine增加、代謝物減少和清除減少相關的不良事件。一項針對三名對Clozapine不耐受且出現遲發性運動障礙(TD)的患者的案例研究,這些患者在低/正常劑量下未使用其他混雜藥物,發現他們的基因型為CYP1A2*1C(2 異型合子(Heterozygous)、1 同型合子(Homozygous)且均無*1F),這是一種低表達等位基因。這些患者淋巴細胞中的CYP1A2-mRNA表達水平低於對照組的1/30[Article:21481946]。另一項針對精神分裂症住院患者的案例系列中,有部分患者接受Clozapine(209名患者中有18名),發現低活性CYP1A2(無誘導劑CBZ、VPA或吸煙,CYP1A2*1D delT或CYP1A2*1F C)與PSP-P和CGI-2分數測量的臨床結果下降相關[Article:25090458]。
一般來說,Clozapine和BMI的PGx研究以及與代謝綜合徵相關的副作用主要集中在PD基因上,並未包括PK基因變異的測量。對於代謝副作用是否與劑量相關存在一些意見分歧[Article:26364648]。一項小型研究(n=17)顯示,低活性變異體CYP1A2 *1C和*1D與較高的血清Clozapine濃度相關,並且在給定劑量的Clozapine上增加了發展胰島素和脂質升高及胰島素抵抗的風險[Article:17503978]。
CYP3A
低CYP3A4表達(以白細胞mRNA水平測量)與高血清Clozapine相關[Article:28340122]。在這些低表達的CYP3A4中,CYP3A5的表達,即CYP3A5*1,導致與CYP3A5*3純合子相比,血清Clozapine減少,且後者未表達功能性CYP3A5 [Article:28340122]。另一項研究顯示,CYP3A5對CYP3A4的影響僅在活動降低的患者中有所貢獻[Article:19593168]。
CYP3A43是CYP3A位點中的另一個基因[Article:20019904],儘管其在Clozapine 途徑中的具體參與尚不清楚,但變異rs680055和rs472660與對Clozapine的反應增加相關[Article:25150845]。
CYP2D6
早期對接受Clozapine的患者的研究顯示,PM和EM基因型與反應之間沒有關聯[Article:7640149]。其他幾項研究得出結論,CYP2D6變異體與Clozapine/n-desmethylclozapine比率或代謝效應並無顯著關聯[Article:7640149][Article:17503978][Article:19593168] [Article:8148222]。FDA藥物仿單未提供支持包含有關CYP2D6變異的文字的參考。
CYP2C19
一項對接受Clozapine的患者的研究顯示,CYP2C19*17/*17基因型,被認為是超快速代謝型(Ultrarapid metabolizer),血清n-desmethylclozapine較高,與糖尿病的較低發病率相關,並且與其精神分裂症症狀的改善增加相關,與CYP2C19*1/*1相比[Article:28664816]。在另一項研究中,弱代謝型(Poor metabolizer)基因型CYP2C19*2/*2與較高的血清Clozapine相關[Article:19593168]。CYP2C19*17/*17與全體隊列中的血清Clozapine或n-desmethylclozapine的顯著變化無關,但在未接受fluvoxamine的小組中卻顯著[Article:19593168]。在一項對91名患者的大型研究中,無論是*2還是*17均未與血清Clozapine的變化相關[Article:23297297]。
運輸蛋白變異
有幾項研究檢查了運輸蛋白中的變異,儘管尚未重複。ABCB1變異rs1045642 AA基因型(3435G>A)與血漿Clozapine增加相關[Article:19593168]。ABCB1的相同基因型rs1045642也與顆粒細胞缺乏症和嗜中性白血球低下症相關[Article:27168101],但另一項研究發現,當該基因型是單倍型的ABCB1的一部分時,可能對顆粒細胞缺乏症具有保護作用[Article:27043126]。一般來說,Clozapine誘導的顆粒細胞缺乏症不被認為是劑量依賴的,但特別是在白細胞中的藥物濃度增加可能是一個因素,並可能受到基因變異的影響。變異rs1045642G在ABCB1和rs212090T在ABCC1與男性但非女性接受Clozapine的體重增加和高血壓風險增加相關[Article:28919802]。
ABCB1 rs7787082 G和rs10248420 A 等位基因在對Clozapine無反應的患者中更常見[Article:22722500]。變異rs2231142在ABCG2中與Clozapine的劑量調整血清谷濃度(Ctrough)增加相關[Article:27932669]。
結論
我們簡要總結了參與Clozapine代謝的候選基因及其在DDI和PGx反應中的作用。Clozapine的PK變異性仍有很大一部分未得到充分解釋。來自DDI的體外和體內實驗的證據並不完全一致[Article:8633698][Article:9928923][Article:9472836][Article:29024893]。這可能有許多原因,包括不同細胞類型中的代謝差異、考慮不同的代謝物以及PGx影響。體外實驗是使用表達的蛋白質或肝臟微粒體進行的,而體內研究可能涉及許多細胞和器官系統。體外系統不允許對CYP1A2或CYP3A4的轉錄調節,這可能是DDI的一個重要部分。 這兩類DDI研究也主要集中在母藥及其兩個主要血清代謝物n-desmethylclozapine和clozapine n-oxide上,對於次要代謝物和途徑在n-desmethylclozapine下游的相關性知之甚少。很少有研究考慮PGx變異對DDI的影響。
目前,PGx證據非常分散,且很少有關聯被重複,但CYP1A2、CYP2C19和CYP3A家族基因都是良好的候選者。CYP2D6變異對Clozapine的PGx影響缺乏證據,儘管DDI的研究顯示CYP2D6參與Clozapine的PK,並且可能在生成次要代謝物中發揮作用[Article:7891353]。 [Article:11147928][Article:8195452][Article:28291036]。需要更大規模的體內研究,包括量化環境影響(如咖啡因和吸煙)以及多個PGx候選基因的機制。雖然一項小型研究得出吸煙對Clozapine/n-desmethylclozapine血漿濃度的影響與每天吸煙的香煙數量無關(>20、11-20、6-10、<=5),但該研究僅檢查了45名吸煙者[Article:19593168]。最近一項將咖啡因代謝和863名健康個體(包括385名吸煙者)的吸煙數據結合的研究,模型化Clozapine的代謝,顯示香煙數量與清除率相關。該模型能夠預測Clozapine的清除率,與已發表的每日香煙數量範圍(>20、11-20、6-10、<=5)一致[Article:22258279]。生成這樣的模型的困難在於考慮咖啡消費可能對CYP1A2表達的誘導影響[Article:18157525]。被動吸煙也可能需要考慮,因為有證據表明它影響了苯乙烯的代謝,苯乙烯是CYP1A2的另一種底物。被動吸煙者的苯乙烯代謝介於吸煙者和非吸煙者之間[Article:9712458]。此外,患者的居住狀況變化應在任何預測模型中考慮,因為從家庭護理、醫院急診護理和住院設施護理的變化可能影響吸煙行為。預測護理安排變化的影響大小的能力可能有助於避免不良事件。還需考慮其他誘導劑/抑制劑,包括環境毒素(如果患者處於不穩定的居住情況下可能會遇到)、處方藥、濫用藥物、替代藥物、酒精和飲食。有證據表明,炎症誘導的下調CPY1A2和CYP3A4導致從EM基因型轉變為PM表現型 [Article:25519488]。
需要更多有關CYP3A5和CYP3A43的貢獻數據。雖然基因變異似乎對CYP3A4的影響有限,但有些變異對CYP3A5和CYP3A43的影響很大[Article:28340122][Article:25150845]。此外,運輸蛋白變異在肝臟和血腦屏障的影響需要更一致的檢查。將所有這些因素結合到演算法中,連同PD候選基因的因素,是最終目標。