CPIC 指南註解:aminosalicylic acid、chloramphenicol、chloroquine、ciprofloxacin、dimercaprol、doxorubicin、furazolidone、glyburide、hydroxychloroquine、mafenide、nalidixic acid、norfloxacin、ofloxacin、phenazopyridine、quinine、sulfadiazine、sulfamethazine、sulfamethoxazole、sulfamethoxazole / trimethoprim、sulfanilamide、sulfasalazine、sulfisoxazole、tolbutamide、vitamin c、Vitamin K and analogues 與 G6PD 基因
摘要
對於具有G6PD 缺乏或G6PD 可變型(Variable) 表現型的患者,沒有理由避免使用低至無風險的藥物。在患有慢性非球形紅細胞溶血性貧血(chronic non-spherocytic hemolytic anemia, CNSHA)的G6PD 缺乏患者中,應謹慎使用低至無風險的藥物,並建議密切監測慢性溶血的急性加重。然而,CPIC不建議基於G6PD基因型改變低至無風險藥物的處方行為。
註釋
此註釋基於擴展的CPIC®指引,針對G6PD基因型的藥物使用。CPIC作者評估了攜帶G6PD變異的患者使用各種藥物的現有證據。
2022年9月
-
擴展的CPIC®指引,針對G6PD基因型的藥物使用已在《Clinical Pharmacology and Therapeutics》期刊中發表。作為該指引的一部分,作者將藥物按其在G6PD缺乏症患者中引起急性溶血性貧血(AHA)的風險進行分類。4-aminosalicylic acid, chloramphenicol, chloroquine, ciprofloxacin, dimercaprol, doxorubicin, furazolidone, glyburide, hydroxychloroquine, mafenide, nalidixic acid, norfloxacin, ofloxacin, phenazopyridine, quinine, sulfadiazine, sulfadimidine, sulfamethoxazole, sulfanilamide, sulfasalazine, sulfisoxazole, tolbutamide, vitamin C和vitamin K被分類為低至無風險藥物,如指引發表的表2所示。CPIC不建議基於基因型改變低至無風險藥物的處方行為,並且G6PD/低至無風險藥物基因/藥物對被CPIC分配為C級最終狀態。
-
這些指引適用於:
- 成人患者
- 兒科患者
-
指引摘錄:
- 「在此CPIC指引中增加了一個步驟,將藥物分為三組:在G6PD缺乏症存在下可被視為高風險的藥物(因此通常應避免),在G6PD缺乏症中被視為中等風險的藥物(因此應謹慎使用),以及可被視為低至無風險的藥物(在G6PD缺乏症患者中不會增加AHA風險)。」
- 「為了將藥物分配到風險組,作者不僅考慮了主要同行評審文獻中的證據強度,還考慮了藥物使用頻率、監管機構警告的存在以及是否存在可能產生活性氧物質並在G6PD缺乏症中促成溶血的機制(補充,分配風險等級)」
- 「Sulfamethoxazole(低至無風險藥物) 在G6PD缺乏症中sulfamethoxazole的風險類別。 Sulfamethoxazole被認為屬於低至無風險類別,因為將其與AHA聯繫起來的證據較弱(主要是病例報告,常常因感染、蠶豆或其他藥物的存在而混淆),且有幾項研究顯示在G6PD缺乏症中安全使用。一項研究報告顯示在10名缺乏嬰兒中未見此藥引起AHA。儘管有一些監管機構的警告,但大多數較弱。此外,sulfamethoxazole被廣泛使用,進一步證明缺乏支持顯著風險的研究。」
-
下載並閱讀:
- 擴展的藥物基因體學臨床應用聯盟(CPIC)指引,針對G6PD基因型的藥物使用
- 2022補充
- G6PD 基因資訊表
- 藥物資源映射
- 4-aminosalicylic acid
- chloramphenicol
- chloroquine
- ciprofloxacin
- dimercaprol
- doxorubicin
- furazolidone
- glyburide
- hydroxychloroquine
- mafenide
- nalidixic acid
- norfloxacin
- ofloxacin
- phenazopyridine
- quinine
- sulfadiazine
- sulfadimidine
- sulfamethoxazole/trimethoprim
- sulfanilamide
- sulfasalazine
- sulfisoxazole
- tolbutamide
- vitamin C
- vitamin K
- 測試前後警示
- 4-aminosalicylic acid
- chloramphenicol
- chloroquine
- ciprofloxacin
- dimercaprol
- doxorubicin
- furazolidone
- glyburide
- hydroxychloroquine
- mafenide
- nalidixic acid
- norfloxacin
- ofloxacin
- phenazopyridine
- quinine
- sulfadiazine
- sulfadimidine
- sulfamethoxazole/trimethoprim
- sulfanilamide
- sulfasalazine
- sulfisoxazole
- tolbutamide
- vitamin C
- vitamin K
- 低至無風險藥物流程圖
表1:低至無風險藥物在G6PD 表現型中的推薦治療使用
改編自指引的表1、2和6
| 預測表現型 | 基因型a | 基因型範例b | 影響 | 治療建議 | 建議等級c | 考量 |
|---|---|---|---|---|---|---|
| 正常 | 攜帶一個X染色體,帶有非缺乏(IV類)等位基因 或攜帶兩個非缺乏(IV類)等位基因者 |
B, Sao Boria, IV B/B, B/Sao Boria, B/A, IV/IV |
急性溶血性貧血的低至無風險 | 基於G6PD狀態,無理由避免低至無風險藥物 | 強烈建議 | |
| 缺乏 | 攜帶一個X染色體,帶有缺乏(II-III類)等位基因 或攜帶兩個缺乏(II-III類)等位基因者或一個I類等位基因和一個II或III類等位基因者 |
A-, Orissa, Kalyan-Kerala, Mediterranean, Canton, Chatham, II, III A-/A-, A-/Orissa, Orissa/ Kalyan-Kerala, Mediterranean/ Mediterranean, Chatham /Mediterranean, Canton/ Viangchan, II/II, II/III, III/III, I/II, I/III |
急性溶血性貧血的低至無風險 | 基於G6PD狀態,在標準劑量下無理由避免低至無風險藥物 | chloramphenicol, chloroquine, hydroxychloroquine, vitamin C, vitamin K: 中等建議 所有其他低至無風險藥物: 可選建議 |
對於高於正常劑量的情況,並在感染或其他氧化壓力的情況下,包括同時使用多種中等和低至無風險藥物時,可能需要更密切的監測。 |
| 嚴重缺乏合併慢性非球形紅細胞溶血性貧血(Deficient with CNSHA) | 攜帶一個X染色體,帶有缺乏(I類)等位基因 或攜帶兩個缺乏(I類)等位基因者d |
Bangkok, Villeurbanne, I Bangkok/Bangkok, Bangkok/Villeurbanne, I/I |
慢性溶血性貧血急性加重的高風險 | 在此組中應謹慎使用所有藥物;如果使用藥物,建議密切監測慢性溶血性貧血的急性加重。 | 可選建議 | 在G6PD 嚴重缺乏合併慢性非球形紅細胞溶血性貧血(Deficient with CNSHA) 表現型患者中數據不足,但由於潛在的病理生理學賦予慢性溶血性貧血急性加重的高風險,應仔細權衡使用任何藥物的風險與益處。 |
| 可變型(Variable)e | 攜帶一個非缺乏(IV類)等位基因和一個缺乏(I-III類)等位基因者 | B /Bangkok, B/Mediterranean, B/A-, IV/I, IV/II, IV/III | 急性溶血性貧血的低至無風險 | 基於G6PD狀態,在標準劑量下無理由避免低至無風險藥物 | 中等建議 | 由於X 染色體聯鎖嵌合現象(X-linked mosaicism),攜帶多於一個X染色體的個體(例如,女性,克氏綜合症患者)和異型合子(Heterozygous)一個非缺乏(IV類)和一個缺乏(I–III類)等位基因者可能顯示正常或缺乏 表現型。 |
| 無法判定 | 攜帶至少一個等位基因具有功能尚未確定 | Dagua B/Dagua |
急性溶血性貧血的未知風險 | 為確定G6PD狀態,必須測量酶活性。藥物使用應根據基於活性表現型的建議進行指導。 | 中等建議 |
CNSHA:慢性非球形紅細胞溶血性貧血
a WHO分類來自[文章:22293322], 其他細節來自[文章:4963040]。I類等位基因極為罕見;II類和III類等位基因之間的區分不明顯。幾乎所有患者都攜帶II、III或IV類等位基因。
b 由於G6PD 等位基因的數量龐大,除了此處給出的範例外,還可能存在其他基因型;請參閱G6PD 等位基因定義表以獲取更全面的等位基因和G6PD 等位基因功能表以了解其分配的功能(WHO類別)。請注意,一些實驗室使用“B 等位基因”的標示來指示不攜帶已知I-III類變異的等位基因。可參考G6PD頻率表以了解G6PD 等位基因在主要生物地理群體中的頻率。
c 評分方案在指引補充的建議強度部分中描述。
d 此類基因型從未見過,推測極為罕見。
e 由於X 染色體聯鎖嵌合現象(X-linked mosaicism),攜帶一個非缺乏(IV類)和一個缺乏(I-III 等位基因)等位基因的個體(通常為女性)可能顯示正常或缺乏 表現型。因此,很難預測這些個體的表現型(見補充,G6PD異合子)。
Uricosurics 途徑, 藥效學
概括
一個風格化的腎近端小管細胞圖示,顯示尿酸排泄藥物在防止人類腎臟重新吸收 uric acid 的作用。

描述
背景
高尿酸血症是uric acid的積累,可能導致痛風和腎衰竭等病理狀況,並且是腫瘤溶解綜合症(TLS)的相關症狀[Articles:20562597, 12646938]。在生理pH下,大多數uric acid以尿酸陰離子的形式存在[Article:20613716。我們在此使用「uric acid」這一術語來涵蓋尿酸。治療或預防高尿酸血症的藥物包括那些抑制uric acid形成的藥物、促進排泄的藥物以及防止再吸收的藥物。本文討論了尿酸排泄藥物在防止人類腎臟中uric acid再吸收的作用,以及參與此過程的基因(見上圖)。uric acid-降低藥物途徑,PD]涵蓋了那些抑制uric acid形成或直接作用於uric acid以增加分泌的藥物。
藥效學
大多數uric acid通過腎臟排泄,然而大約90%的比例被腎近端小管中表達的轉運蛋白再吸收[Article:20613716]。尿酸排泄藥物通過針對轉運蛋白來抑制腎近端小管中uric acid的再吸收(見上圖)[Articles:20562597, 22665944]。許多轉運蛋白在腎近端小管的腔面頂膜和間質基底膜的uric acid的分泌和再吸收中具有假定的作用;然而,尿酸運輸的機制尚未明確[Articles:22359229, 22038265]。最近的基因分析,如全基因組關聯研究(GWAS),已經確定了與uric acid水平或痛風相關的新基因變異,並幫助闡明哪些蛋白質可能在uric acid的運輸和人類的血漿水平中起重要作用[Articles:20613716, 22359229, 22038265, 22945592, 18606621, 18327257, 23263486, 22797727]。目前的模型涉及一組蛋白質:尿酸或uric acid的「運輸體複合體」[Articles:22038265, 22359229, 22945592]。運輸體複合體的轉運蛋白參與其他化合物的運輸,但在此我們專注於uric acid。
已知的uric acid轉運蛋白
URAT1 (SLC22A12)是一種轉運蛋白,位於腎近端小管(上皮)細胞的頂面,主要負責從腔內攝取uric acid [Articles:12024214, 20613716, 22359229, 22945592, 21272127]。在腎低尿酸血症患者中識別出的SLC22A12基因的變異缺乏uric acid的轉運活性,或與野生型URAT1相比顯示出顯著降低的活性in vitro [Articles:12024214, 14694169]。同樣在腎近端小管細胞的頂膜上表達的轉運蛋白,參與uric acid向腎小管濾液/腔的分泌,由ABCG2(BCRP)、SLC17A1(NPT1)和SLC17A3(NPT4)編碼[Articles:20613716, 22359229, 22945592]。GLUT9 (SLC2A9)在基底膜上表達,主要負責將uric acid運輸到間質和血液中[Articles:20613716, 22359229, 22945592]。證據表明存在兩種GLUT9的亞型 - GLUT-9a(亞型1)位於基底側,GLUT-9b(亞型2或GLUT9N)位於頂側,儘管後者的作用仍不明確[Articles:20613716, 22945592, 22359229]。in vitro,兩種亞型均能在穩定表達的細胞中運輸uric acid [Article:18842065]。
驗證其在uric acid運輸中的作用,最近兩項不同的綜合分析(Meta-analysis)研究(均在超過28,000名歐洲血統/白人族裔的個體中進行)發現與血清uric acid水平相關的SNP位於包含SLC22A12、SLC2A9、SLC17A1、SLC17A3和ABCG2基因的位點,以及其他新位點[Articles:19503597, 20884846]。一項GWAS的meta分析檢查了四項表現型與腎功能相關的研究,驗證了SLC2A9、ABCG2和SLC22A12位點與33,074名東亞個體的uric acid濃度相關[Article:22797727]。GWAS的meta分析涵蓋了來自全球尿酸遺傳學聯盟(GUGC)的超過140,000名歐洲血統的個體,確定了10個已知位點和16個與血清尿酸濃度在全基因組顯著性相關的新位點[Article:23263486]。這些位點解釋了尿酸濃度變異的7%,其中SLC2A9和ABCG2位點貢獻了3.4%的變異[Article:23263486]。與尿酸濃度相關的17個位點也與痛風相關[Article:23263486],這在之前的研究中已顯示出與SLC2A9和ABCG2位點相關[Article:20884846]。許多與SNP濃度相關的uric acid位點在歐洲分析中也與非裔美國人(n=5,820)、日本人(n=15,286)和印度祖源個體(n=8,340)的隊列相關,儘管觀察到差異,可能是由於四個不同人群之間的等位基因頻率變異所致[Article:23263486]。有趣的是,最近的GWAS 綜合分析(Meta-analysis)報告中與uric acid濃度相關的新位點包括編碼轉錄因子的基因和與葡萄糖穩態及腎功能相關的基因,揭示了可能的新藥物發現途徑,以降低尿酸水平並預防腎病[Articles:22797727, 23263486, 19503597, 20884846]。
潛在的uric acid轉運蛋白
上圖中陰影部分顯示的幾個其他基因在人體腎近端小管中可能在uric acid運輸中發揮作用(以虛線箭頭表示)。這些基因均有in vitro的證據,其中一些也在與血清uric acid濃度和/或痛風相關的GWAS中被識別:
- SLC22A6 (OAT1)和SLC22A8 (OAT3)在基底膜上表達,in vitro的研究顯示它們可以攝取uric acid,而基因敲除小鼠的uric acid分泌減少[Articles:22359229, 22945592, 17674156]。in vitro,人類OAT1對uric acid的攝取受到probenecid和benzbromarone的抑制[Article:12472777]。
- SLC22A11 (OAT4)位於頂膜上,並能在in vitro中攝取uric acid [Articles:22359229, 20613716, 22945592, 15037815, 17674156]。SLC22A11位於SLC22A12基因的上游,並在綜合分析(Meta-analysis)的GWAS中識別出幾個與血清uric acid水平相關的SNP [Articles:19503597, 20884846]。由於SLC22A12和SLC22A11在第11號染色體上彼此接近,因此可能難以區分與此位點的SNP的個別信號[Article:20884846]。
- SLC22A13 (OAT10)在腎近端小管細胞的頂側表達,並能在in vitro中攝取uric acid,此過程受到probenecid的顯著抑制[Articles:21103968, 22038265, 18411268]。
- LGALS9(galectin 9,UAT)被認為在腎近端小管細胞的基底側和頂側運輸uric acid中發揮作用[Article:22359229]。
- ABCC4 (MRP4)在腎近端小管細胞的頂側表達,作為ATP-依賴的uric acid離子泵,可能參與uric acid的分泌,然而目前尚無人類或動物數據確認此角色[Articles:22359229, 20613716]。
- PDZK1編碼一種接頭蛋白,位於接近頂側的位置,與包括URAT1、OAT4和NPT1的轉運蛋白相互作用,可能促進uric acid的有效運輸[Articles:22359229, 22945592]。在一項GWAS的meta分析中,位於PDZK1基因上游的SNP與血清uric acid水平相關[Article:19503597]。
尿酸排泄藥物
probenecid最初是為了抑制腎臟對青黴素的分泌而開發的,後來發現其對痛風患者的益處[Article:23318701]。它主要通過抑制URAT1(SLC22A12)轉運蛋白的活性來防止有機陰離子如uric acid的再吸收[Articles:12024214, 22359229, 22665944, 23318701, 21272127]。in vitro,probenecid似乎對GLUT9(SLC2A9)在1mM的有效藥理濃度下沒有影響[Article:18842065]。probenecid也可能通過作用於OAT1(SLC22A6)、OAT4(SLC22A11)和OAT10來防止uric acid的再吸收[Articles:23318701, 12472777, 18411268]。因此,probenecid可以改變同時服用藥物的清除率,並影響咖啡因的代謝[Articles:11823755, 4020675, 23436258, 17725176]。probenecid對SLC22A8(OAT3)的抑制可能意味著它是一種潛在的治療策略,用於治療流感A感染[Article:23129053]。benzbromarone通過URAT1(SLC22A12)和GLUT-9(SLC2A9)來抑制uric acid的運輸in vitro,儘管在治療劑量下對GLUT-9的作用可能是微小的[Articles:23318701, 22945592, 18670416, 18636784, 12024214, 21272127, 18842065]。它還顯示出對uric acid的攝取的作用,通過OAT1(SLC22A6) in vitro [Article:12472777]。sulfinpyrazone通過uric acid的攝取來抑制URAT1(SLC22A12) in vitro,並在體內降低血清uric acid [Articles:12024214, 1948784, 668783]。它還抑制其他轉運蛋白的活性,包括ABCC1、ABCC2和ABCC10 [Articles:18445659, 14569069, 9685354]。
其他具有尿酸排泄特性的藥物
用於高血壓治療的losartan是一種獨特的血管緊張素II受體拮抗劑,因為它還增加uric acid的分泌,並通過抑制腎近端小管中的再吸收顯著降低血漿水平,針對轉運蛋白URAT1(SLC22A12) [Articles:8743498, 18670416, 12024214]。tranilast是一種抗炎藥,但也通過抑制URAT1和GLUT9顯示出尿酸排泄特性,因此這些綜合效果使其成為潛在的痛風治療藥物[Article:23318701]。vitamin c被認為通過抑制uric acid的再吸收來作為尿酸排泄藥物,URAT1(SLC22A12) [Article:23253231]。研究顯示,500mg/每日的劑量可能降低血清uric acid,而劑量超過1000mg/每日可能降低痛風的風險[Article:22198943]。水楊酸鹽/salicylic acid和indomethacin也通過抑制uric acid的攝取來影響URAT1(SLC22A12) in vitro [Articles:12024214, 21272127]。
藥物基因體學 (PGx)
除了影響血漿uric acid水平和痛風風險外,轉運蛋白基因中的多態性也可能影響Uricosurics抑制再吸收的療效。然而,目前針對Uricosurics的PGx研究較少。以下是通過廣泛文獻檢索發現的與Uricosurics的基因關聯。上圖顯示了參與uric acid運輸調控的基因,突顯了潛在的新藥物靶點和未來藥物基因體學研究的潛在基因。參與這些藥物代謝的基因中的多態性也可能影響其毒性和療效。
SLC22A12 (URAT1)
在低尿酸血症患者中已識別出多種SLC22A12的變異,其中一些顯示出降低的uric acid攝取in vitro [Articles:14694169, 12024214, 18492088, 15327384]。有一些證據表明,這些變異可能影響對作用於URAT1的尿酸排泄和抗尿酸排泄藥物的反應。在高血壓患者(無低尿酸血症)中,具有野生型SLC22A12的患者在benzbromarone或losartan治療下,uric acid的清除率(Cur)/肌酐清除率(Ccr)比率顯著增加(表明血清uric acid的降低)。在高血壓和低尿酸血症患者中,對同型合子(Homozygous)或複合異形合子(Compound heterozygous)的SLC22A12基因變異進行檢測後,benzbromarone或losartan的治療對Cur/Ccr比率沒有影響(表明對血清uric acid水平沒有影響)。losartan仍能在這些患者中維持其降壓效果[Article:18670416。這些變異包括rs121907896 G269A(Arg90His)和rs121907892] G774A Trp258Ter(c.NM_144585.2);這兩者均在特發性腎低尿酸血症患者中被識別,與降低的uric acid攝取in vitro和與野生型相比,受試者的血清uric acid殘留水平較低[Articles:12024214, 15327384, 14694169]。這些低尿酸血症患者對benzbromarone的缺乏反應,與SLC22A12變異的患者中觀察到的另一項研究結果相符,該研究也觀察到吡嗪酰胺的抗尿酸排泄活性受到影響[Article:14694169]。這可能對低尿酸血症患者的治療有影響。
CYP2C9
由於報告顯示致命的肝毒性,benzbromarone已被其主要製造商撤回市場,儘管這被一些人認為是過早的,並且在全球幾個國家的其他製藥公司仍然可以獲得[Articles:18636784, 22933344]。基因可能是影響毒性風險的因素。benzbromarone由CYP2C9代謝形成活性代謝物6-羥基benzbromarone(對URAT1uric acid的攝取具有抑制活性)和由CYP3A4代謝形成1'-羥基benzbromarone [Articles:22933344, 18636784, 18020424, 21272127]。6-羥基benzbromarone進一步被CYP2C9和CYP1A2代謝為5,6-二羥基benzbromarone [Article:22933344]。在20名健康個體的小型研究中,發現具有CYP2C9*3/*3基因型的個體相比於具有*1/*1或*1/*3基因型的個體,代謝和清除benzbromarone的能力降低,且在具有*1/*3基因型的患者中,6-羥基benzbromarone的消除半衰期顯著高於*1/*1 [Article:20962433]。CYP2C9基因型是否對benzbromarone 藥效學有臨床相關的影響尚不清楚;該研究未觀察到uric acid的排泄或血漿濃度的差異[Article:20962433]。在CYP2C9中的多態性導致benzbromarone的血漿水平升高和對6-羥基benzbromarone的代謝降低,可能會導致毒性:benzbromarone和1'-羥基benzbromarone均顯示出細胞毒性效應in vitro [Article:23229783],而5,6-二羥基benzbromarone也可能導致毒性[Article:18020424]。CYP2C9依賴的連續氧化形成5,6-兒茶酚,具有形成反應性醌的潛力,這一過程由形成谷胱甘肽加合物所支持in vitro [Article:18020424]。這一反應特徵在幾種其他肝毒性藥物中是典型的。建議在苯溴馬林引起的肝毒性病例中進行CYP2C9基因分型[Article:18636784],以開始收集更多證據,了解CYP2C9基因型與benzbromarone毒性之間是否存在關聯。CYP2C9中的多態性也可能改變藥物交互作用。benzbromarone通過CYP2C9抑制氟比洛芬(抗炎藥)的代謝,然而在CYP2C9.3的存在下,氟比洛芬的代謝被激活[Article:15955872]。
ABCB1和ABCC2
迄今為止,尚未找到任何藥物基因體學研究檢查針對uric acid的基因變異對分泌的直接影響。然而,有幾項研究檢查了其他藥物,使用probenecid作為抑制劑。在一項小型研究中,probenecid降低了藥物二氯西林的清除率和分佈,並且在共同給藥時,SNP在ABCB1的外顯子26中顯示出影響二氯西林尿液排泄的作用[Article:18576903],這表明可能存在基因對probenecid作為抑制劑的療效的影響,但仍需進一步研究。probenecid抑制通過野生型MRP2蛋白的羧基熒光素的外排in vitro [Article:11477083]。兩個ABCC2基因的變異(編碼MRP2),與Dubin-Johnson綜合症相關,導致轉運活性受損(R11150H 3449G>A,3517A>T I1173F) [Article:11477083],因此這些變異可能影響probenecid對此轉運蛋白的作用。
UGT1A1
sulfinpyrazone的內源性清除率通過UGT1A9的葡萄糖醛酸化顯著降低,這在表達UGT1A9變異Met33Thr(rs72551330 等位基因 C)中顯示出來,與Met33(等位基因 T)相比[Article:22981363]。
總結
由於約90%的uric acid在體內被腎近端小管再吸收,針對參與再吸收的轉運蛋白的藥物已被開發用於治療高尿酸血症/痛風。涉及的機制尚待闡明,但在此我們提供已知和潛在的參與uric acid運輸的基因,這些基因在腎近端小管細胞中被尿酸排泄藥物靶向。進一步研究這些基因中的多態性對這些藥物尿酸排泄作用的影響,可能會提供對治療效果或新藥開發靶點的見解。
Oxidative Stress Regulatory 途徑 (紅血球)
概括
一個簡化的圖示,顯示幾種防止紅血球氧化壓力的調控機制,其中許多機制需要來自於 Pentose Phosphate 途徑 的 NADPH。
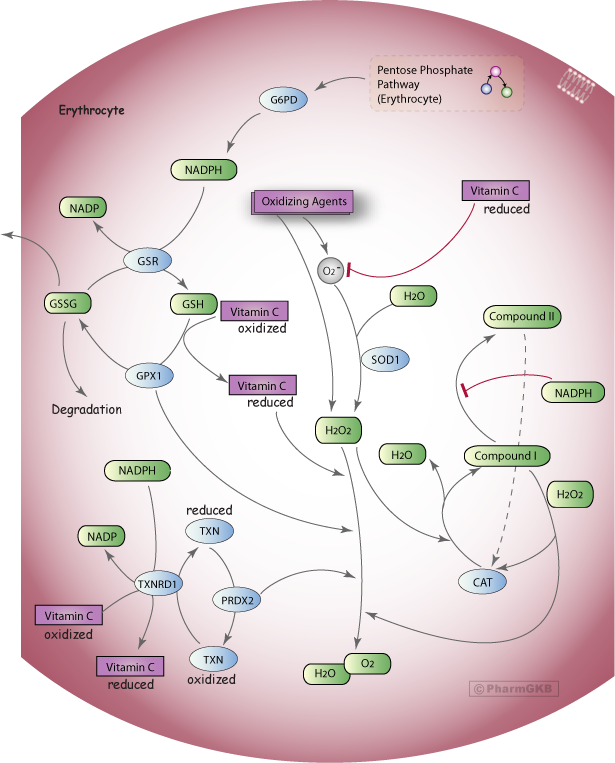
描述
反應性氧物種的釋放 (ROS),例如超氧化物 (O2-) 和/或過氧化物(例如氫過氧化物,H2O2),可以被外源性氧化劑如藥物或其代謝物所觸發 [Articles:2633878, 10533013]。例如,H2O2 是藥物 rasburicase 將尿酸轉化為尿囊素的副產物 [Articles:12646938, 16597166]。紅血球 (RBCs, erythrocytes) 不斷受到氧化壓力的影響,這是由於它們作為氧運輸者的角色以及在循環中接觸外源性物質 [Articles:15862084, 10533013]。Oxidative Stress Regulatory 途徑 專注於一些需要 NADPH 來中和 ROS 的機制,例如 O2- 和 H2O2。
O2- 可以通過超氧化物歧化酶 (SOD1) 轉化為 H2O2 [Articles:20350285, 2633878]。谷胱甘肽還原酶 (GSR) 利用 NADPH 將氧化型谷胱甘肽 (GSSG) 轉化為還原型谷胱甘肽 (GSH) [Articles:18177777, 16204390, 21376665, 17611006, 10657232]。然後,GSH 被谷胱甘肽過氧化酶 (GPX1) 氧化回 GSSG,這是一個循環反應,用於將 2H2O2 中和為 2H20 和 O2 [Articles:18177777, 16204390, 21376665, 17611006, 2633878]。GSH 也通過防止和逆轉氧化來保護血紅蛋白,這種氧化會導致球蛋白鏈之間的二硫化物交聯,並扭曲血紅蛋白結構,可能導致“海因茲小體”的沉澱 [Article:15862084]。具有正常 G6PD 酶活性的 RBCs 其 PPP 活性和 GSH 水準較高,與 G6PD 缺乏 RBCs 相比,然而 缺乏 細胞在正常條件下可以應對低水平的可用 NADPH [Articles:4154443, 2633878]。當氧化壓力發生時,G6PD “正常” RBCs 通過增強 PPP 活性來維持 GSH 水準,而在 G6PD 缺乏 細胞中,PPP 保持在最低容量,GSH 水準下降 [Article:4154443]。由於 PPP 在 G6PD 缺乏 RBCs 中已接近正常條件下可獲得的最大活性速率,因此它們無法應對氧化壓力,並且更容易受到氧化壓力觸發的溶解,這可能導致溶血性貧血 [Articles:4154443, 2633878]。
另一個需要 NADPH 的關鍵系統涉及氧化型硫氧還蛋白 (TXN) 轉化為還原型,這一過程由硫氧還蛋白還原酶 (TXNRD1) 催化,還原的 TXN 隨後被酶過氧化物還原酶用作電子供體,以中和 H2O2 [Articles:18479207, 17611006, 10657232]。在 RBCs 中,過氧化物還原酶 2 (Prx 2, PRDX2) 是最豐富的同種型,並且通過與血紅蛋白結合和穩定血紅蛋白發揮保護作用 [Articles:18479207, 22960070]。PRDX2 的重要性在於缺失小鼠中顯示出形態學 RBC 缺陷,例如海因茲小體,並且在接觸 H2O2 血液樣本時,缺失小鼠的樣本相比於野生型樣本顯示出增強的 MetHb 形成 [Article:12586629]。
GSH 和 TXNRD1 都可以將完全氧化的 vitamin c(去氫抗壞血酸)轉化為其還原型(抗壞血酸),這可以進一步向 O2-、H2O2 和氧自由基捐贈電子和氫 [Articles:10657232, 9667500, 9405334, 11687303]。由於人類無法合成 vitamin c,因此從氧化狀態回收它對於維持 RBC 和抗氧化形式的血漿水準至關重要 [Articles:10657232, 9667500, 9405334]。
CAT酶 (CAT) 也中和 H2O2,涉及形成不同狀態的酶的反應步驟 - NADPH 不是 CAT酶功能活性所必需的,而是防止形成酶的非活性狀態 [Article:17158050]。靜止的 CAT酶(鐵CAT酶)與 H2O2 的第一次反應形成化合物 I 和 H2O,然後與進一步的 H2O2 分子反應使 CAT酶返回靜止狀態並釋放 H2O 和 O2 [Articles:17158050, 22516655]。化合物 I 的還原也可以形成非活性的化合物 II 狀態,該狀態會慢慢自發地恢復為 CAT酶 [Articles:17158050, 22516655。化合物 II 的形成可以通過 NADPH 的產生來防止,該產物由 G6PD 從葡萄糖-6-磷酸生成(見 五碳磷酸 途徑(紅血球)] [Articles:3805001, 8704218, 17158050]。證據表明,NADPH 也可能在延長此狀態的 CAT酶的條件下,將化合物 I 還原回 CAT酶 [Article:17158050]。