CPIC 指南註解:fluoxetine 與 CYP2D6 基因
摘要
目前有無建議用於根據CYP2D6基因型進行fluoxetine劑量調整。
註釋
此註釋基於CPIC® 指南,針對CYP2D6、CYP2C19、CYP2B6、SLC6A4和HTR2A基因型與血清素再攝取抑制劑抗抑鬱藥。
2023年2月
CPIC® 指南的作者針對CYP2D6、CYP2C19、CYP2B6、SLC6A4和HTR2A基因型與血清素再攝取抑制劑抗抑鬱藥,評估了攜帶CYP2D6和CYP2C19變異的患者使用fluoxetine的現有證據。
-
指南摘錄:
- 「已證明CYP2D6超快速代謝者(UMs)或慢代謝者(PMs)具有顯著不同的母藥與代謝物比率(見表S1),但fluoxetine加norfluoxetine的血漿總濃度可能不會因CYP2D6代謝者組別而顯著變化(43)。」
- 「關於CYP2D6 表現型狀態如何影響fluoxetine加norfluoxetine濃度總和隨時間變化的數據很少,或CYP2D6 表現型狀態引起的fluoxetine與norfluoxetine濃度不平衡是否影響患者結果或安全性。因此,未提供基於基因的fluoxetine劑量建議。」
-
下載並閱讀:
表1:基於CYP2D6 表現型的fluoxetine劑量建議
改編自指南/補充資料的表1和S6。
| 表現型 | 活性分數 範圍 |
活性分數 | 範例 | 影響 | 治療 建議 |
建議 分類a |
|---|---|---|---|---|---|---|
| CYP2D6 超快速代謝型(Ultrarapid metabolizer) | >2.25 | >2.25 | *1/*1xN, *1/*2xN, *2/*2xNc | fluoxetine的代謝增加,fluoxetine:norfluoxetine比率降低,與正常代謝型(Normal metabolizer)相比。缺乏支持fluoxetine:norfluoxetine比率降低的臨床影響的證據。由於fluoxetine和norfluoxetine對CYP2D6的抑制,超快速代謝型(Ultrarapid metabolizer)轉變為正常、中間或弱代謝型(Poor metabolizer)的程度尚不清楚。 | 基於基因型對fluoxetine不建議採取行動,因為關於對療效或副作用影響的證據有限。 | 無建議 |
| CYP2D6 正常代謝型(Normal metabolizer) | 1.25 <= x <= 2.25 | 1.25 1.5 1.75 2.0 2.25 |
*1/*10 *1/*41, *1/*9 *10/*41x3 *1/*1, *1/*2 *2x2/*10 |
正常代謝。由於fluoxetine和norfluoxetine對CYP2D6的抑制,正常代謝型(Normal metabolizer)轉變為中間或弱代謝型(Poor metabolizer)的程度尚不清楚。 | 以建議的起始劑量開始治療。 | 強烈建議 |
| CYP2D6 中間代謝型(Intermediate metabolizer) | 0 < x < 1.25 | 0.25 0.5 0.75 1.0 |
*4/*10 *4/*41, *10/*10 *10/*41 *41/*41, *1/*5 |
fluoxetine的代謝減少,fluoxetine:norfluoxetine比率增加,但與正常代謝型(Normal metabolizer)相比,總活性對映異構體濃度相似。缺乏支持fluoxetine:norfluoxetine比率增加的臨床影響的證據。由於fluoxetine和norfluoxetine對CYP2D6的抑制,中間代謝型(Intermediate metabolizer)轉變為弱代謝型(Poor metabolizer)的程度尚不清楚。 | 基於基因型對fluoxetine不建議採取行動,因為關於對療效或副作用影響的證據有限。 | 無建議 |
| CYP2D6 弱代謝型(Poor metabolizer) | 0 | 0 | *3/*4, *4/*4, *5/*5, *5/*6 | fluoxetine向活性代謝物的代謝減少,fluoxetine:norfluoxetine比率大幅增加,但與正常代謝型(Normal metabolizer)相比,總活性對映異構體濃度相似。缺乏支持fluoxetine:norfluoxetine比率增加的臨床影響的證據。 | 基於基因型對fluoxetine不建議採取行動,因為關於對療效或副作用影響的證據有限。 | 無建議 |
a 評分方案詳見補充材料。
選擇性血清素再回收抑制劑(SSRI) 途徑, 藥效學
概括
參與血清素合成、釋放、再攝取及介導抗憂鬱劑 選擇性血清素再回收抑制劑(SSRI)在人類大腦中作用的基因。
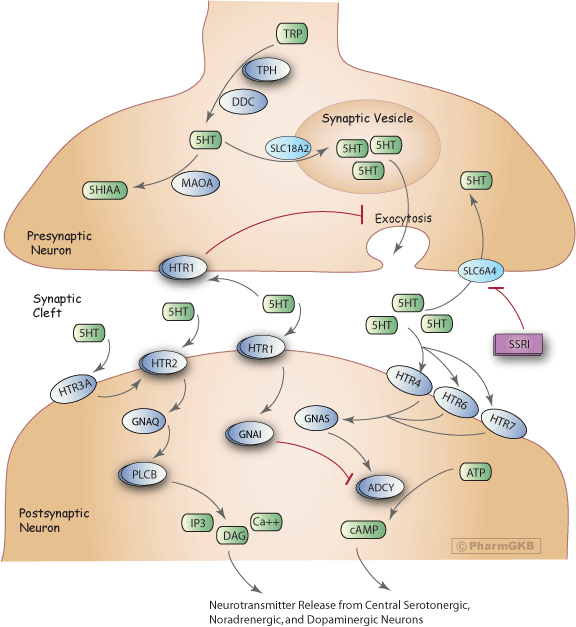
描述
血清素(5-羥色胺,5-HT)是一種神經傳導物質,影響多個過程,包括自主功能、運動活動、荷爾蒙分泌、認知以及與情感、情緒和獎勵相關的複雜過程[Article:17046718]。
在血清素能神經元的末端軸突中,自由色氨酸(TRP)被轉化為5-HT[Article:11590474]。5-HT的合成是一個由色氨酸羥化酶(TPH)和芳香族脫羧酶(DDC)催化的兩步驟過程。TPH是限速酶,存在兩種同工型TPH1和TPH2。在神經組織中,TPH2同工型是主要形式[Articles:12511643, 16581041]。5-HT的攝取進入突觸前儲存囊泡是由囊泡單胺轉運蛋白(SLC18A2)介導的。該轉運蛋白利用囊泡膜上的質子梯度將血清素積累到突觸囊泡中[Article:9665836]。未儲存於囊泡中的5-HT則被單胺氧化酶A(MAOA)降解為5-羥基吲哚乙酸(5-HIAA)。
動作電位刺激突觸前囊泡中血清素的鈣依賴性外排,釋放至突觸間隙,並與突觸後和突觸前受體相互作用。在突觸前側,5-HT激活5-羥色胺(血清素)受體1A(HTR1A)、B(HTR1B)和D(HTR1D),導致5-HT外排的減弱[Article:11590474]。這一反饋迴路調節突觸間隙中的5-HT濃度,因此影響突觸後膜上各種HTR受體亞類的刺激程度[Article:8480026]。長期使用選擇性血清素再回收抑制劑(SSRI)會使這些反饋迴路失去敏感性,從而削弱其對血清素能神經傳導的調節作用[Article:8221701]。突觸後HTR1受體(HTR1A、HTR1B、HTR1D、HTR1E、HTR1F)與HTR2受體亞型(HTR2A、HTR2C)共同作用,通過激活第二信使級聯來介導效應信號[Article:11590474]。在突觸後,訊息傳遞途徑對HTR1受體亞型的主要信號鏈接是通過Gi/o蛋白α亞單位(GNAI)的耦合來實現的。這一相互作用通過抑制腺苷酸環化酶(ADCY)來降低環狀AMP的形成[Article:16896947]。在與5-HT相互作用後,HTR2受體亞群的主要信號鏈接是通過耦合Gq/11蛋白α(GNAQ)來激活磷脂酶C(PLCB)[Article:16896947]。PLCB催化肌醇-1, 4, 5-三磷酸(IP3)和二酸甘油(DAG)的形成[Article:11916537]。突觸後的離子型HTR3受體是一種陽離子特異性配體門控離子通道,並不激活第二信使系統[Article:18466097]。5-HT與此受體的結合通過鈉的內流和鉀的外流使突觸後膜去極化,這被認為會影響HTR2受體的激活[Article:11590474]。HTR4、HTR6和HTR7主要耦合Gs蛋白α(GNAS),導致腺苷酸環化酶的激活,從而增加環狀AMP的水平[Article:11916537]。進一步的操作多樣性得益於多種HTR的剪接和編輯變異的存在、輔助蛋白和伴護蛋白的可能調節,以及它們在GPCR和配體門控通道層面形成同源或異源聚合物的潛力[Article:18571247]。
所有這些第二信使信號在進一步下游反應中的放大導致通過調節鉀通道、幾種蛋白激酶和其他鈣依賴性信號來介導來自中樞血清素能、去甲腎上腺素能和多巴胺能神經元的神經傳導物質釋放。
慢性使用抗抑鬱劑治療已被報導通常會增加腦源性神經生長因子(BDNF)的表達,這是一種依賴於活動的分泌蛋白,對神經網絡的組織和突觸可塑性至關重要[Articles:18433734, 19403460]。
溶質運輸蛋白家族6(神經傳導物質轉運蛋白,血清素),成員4(SLC6A4)負責終止突觸間隙中5-HT的作用。釋放的血清素通過這種整合膜蛋白被運輸回突觸前末端。SLC6A4是Na+/Cl--依賴性轉運蛋白家族的成員[Article:14642278]。
目前存在四大類抗抑鬱藥物:單胺氧化酶抑制劑、選擇性血清素再回收抑制劑(SSRI)、三環化合物和非典型抗抑鬱藥[Article:8221701]。迄今為止,尚未建立針對這些療法的全面假說來解釋其抗抑鬱作用[Article:12726882]。儘管如此,這些藥物都有一個或多個主要的分子靶點以發揮作用。SSRI的分子靶點是SLC6A4,導致抑制突觸前從突觸間隙中5-HT的再攝取。五種SSRI(fluoxetine、fluvoxamine、paroxetine、sertraline和citalopram)在其藥理特徵上有所不同,導致對特定患者的療效或副作用特徵的差異[Articles:10674711, 19370626, 18308785]。SSRI對5-HT再攝取轉運蛋白具有高親和力,對去甲腎上腺素再攝取轉運蛋白具有低親和力,對神經傳導物質受體則具有非常低的親和力。
目前的抗抑鬱療法需要持續治療2-4週才能有效的觀察結果,這表明血清素能和去甲腎上腺素能神經傳導及下游神經適應(例如BDNF受體訊息傳遞途徑)的適應性變化,而不僅僅是突觸單胺水平的升高,才是其治療效果的原因[PMID:19522734, 19481572]。
幾種基因多態性已與治療SSRI反應及不良反應相關,包括SLC6A4、HTR1A、HTR2A、HTR3B、TPH、BDNF和G蛋白β3亞單位的基因變異[Articles:18466097, 19403460, 15111987, 19558256]。
Fluoxetine 途徑, 藥物動力學
概括
參與 fluoxetine 代謝之候選基因示意圖
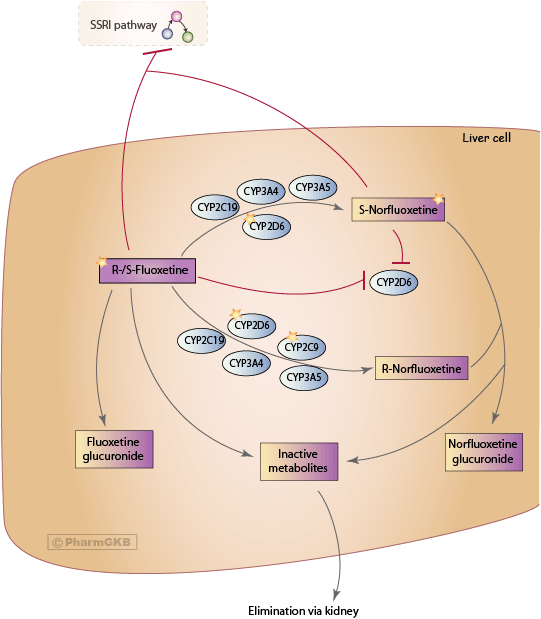
描述
Fluoxetine 是一種 選擇性血清素再回收抑制劑(SSRI)。它已被處方用於治療抑鬱症,並且在治療神經性厭食症和強迫症方面也顯示出療效 [Articles:3501993, 2787123]。Fluoxetine 在口服給藥後幾乎完全被吸收。然而,由於肝臟的首過代謝,其系統性可用性降低。由於其脂溶性特徵,fluoxetine 具有較大的分佈容積,並在多種組織中積累 [Article:8861776]。大腦與血漿的比率為 2.6:1,這低於其他 SSRI [Article:9090339]。
Fluoxetine 在肝臟中被廣泛代謝。唯一已知的活性代謝物,norfluoxetine,是通過 fluoxetine 的去甲基化形成的。fluoxetine 是兩種對映異構體的外消旋混合物。S-fluoxetine 在抑制血清素再攝取方面的效力略高於 R-fluoxetine [Article:7935707]。對於活性代謝物,這一差異更加明顯。S-norfluoxetine 的再攝取阻斷效力約為 R-norfluoxetine 的 20 倍 [Article:1279447]。這四種化合物在其動力學上也存在差異。在幾週的治療後,兩種 S-對映異構體的血漿濃度約為 R-對映異構體濃度的兩倍 [Article:7935707]。
主要的排泄途徑主要是通過氧化代謝和結合,但超過一半的代謝終產物尚不明 [Article:7935707]。fluoxetine 主要通過尿液排泄,未改變或作為 fluoxetine 葡萄糖醛酸鹽排泄的比例低於 10% [Article:2878798]。
來自幾項體外和體內研究的證據表明,CYP2D6、CYP2C19、CYP2C9、CYP3A4 和 CYP3A5 在 R- 和 S-fluoxetine 轉化為其 N-去甲基代謝物的生物轉化中至少部分參與 [Articles:11356927, 10997938]。細胞色素 P450 的同工酶表現出影響其催化活性的遺傳多態性。對不同 CYP2D6 和 CYP2C9 基因型患者的研究結果顯示,CYP2C9 優先催化 R-fluoxetine 的去甲基化,而 S-norfluoxetine 的形成則高度依賴於 CYP2D6 [Articles:11356927, 10208643]。同時,fluoxetine 和 norfluoxetine 的對映異構體是 CYP2D6 媒介反應的抑制劑。因此,在慢性給藥期間,CYP2C19、CYP2C9、CYP3A4 和 CYP3A5 在 fluoxetine 代謝中的影響變得更加重要,因為 CYP2D6 的貢獻因 fluoxetine 和 norfluoxentine 的抑制而減少。
此外,fluoxetine 在體外研究中顯示出對 CYP2C19、CYP2C9 和 CYP3A4 的抑制效力 [Articles:9298519, 9384467, 8703653]。所有這些 CYP 同工酶參與多種藥物的代謝;因此,fluoxetine 和 norfluoxetine 具有改變共同給藥藥物的代謝和 藥物動力學 的潛力。由於 fluoxetine 和 norfluoxentine 對同工酶 CYP2D6 的抑制作用,已報告與 三環抗憂鬱劑(TCA) 和神經安定劑的臨床相關藥物相互作用 [Article:10674711]。