CPIC 指南註解:metoprolol 與 CYP2D6 基因
摘要
CYP2D6 弱代謝型(Poor metabolizer)的患者應以最低建議起始劑量開始使用metoprolol,並仔細調整至臨床效果或指南推薦劑量。或者,可以選擇不同的β-受體阻滯劑。CYP2D6 超快速代謝型(Ultrarapid metabolizer)有無建議。
為特定註釋指定基因型或表現型
註釋
July 2024
《CPIC® 指南》作者針對 beta-blocker 治療與 CYP2D6、ADRB1、ADRB2、ADRA2C、GRK4 和 GRK5 評估了攜帶 CYP2D6 變異的患者使用 metoprolol 的現有證據。
這些指南適用於:
- 成人患者
- 兒科患者
指南摘錄:
支持 CYP2D6 基因型與 metoprolol 暴露和反應相關性的證據包括具有多種健康狀況的參與者(例如,健康、高血壓、心力衰竭等)。因此,可以合理地假設 CYP2D6 變異的藥代動力學效應會在多種適應症中對臨床 metoprolol 反應產生類似影響,並且提供的劑量建議可用於大多數心血管適應症。
建議主要集中在降低 CYP2D6 弱代謝型(Poor metabolizer)s 相關的不良反應風險,這些不良反應源於 metoprolol 系統暴露增加導致的心率和血壓顯著降低。此外,由於這些藥代動力學差異,弱代謝型(Poor metabolizer)s 的最大耐受 metoprolol 劑量可能低於 正常代謝型(Normal metabolizer)s。
雖然證據顯示 CYP2D6 中間代謝型(Intermediate metabolizer)s 的 metoprolol 血漿濃度也高於 正常代謝型(Normal metabolizer)s,但這些效應的幅度似乎小於 弱代謝型(Poor metabolizer)s 所觀察到的,且缺乏足夠的證據來闡明這些較小的藥代動力學差異是否顯著影響臨床反應。
缺乏足夠的證據來確定 CYP2D6 超快速代謝型(Ultrarapid metabolizer)s 是否在 metoprolol 暴露或反應上經歷臨床顯著差異。
重要的是,本指南中提供的任何建議都不應被解釋為會阻止或妨礙 beta-blocker 劑量的上調至最大耐受或指南推薦的水平,例如在射血分數減少的心力衰竭和心肌梗塞(myocardial infarction)後的情況下。
下載並閱讀:
表1:metoprolol 與 CYP2D6 表現型 相關的推薦治療用途
改編自指南的表1和表2
| 表現型a | 活性分數範圍 | 活性分數b | CYP2D6 雙倍型的例子c | 影響d | 建議 | 建議等級e |
|---|---|---|---|---|---|---|
| 超快速代謝型(Ultrarapid metabolizer) | >2.25 | >2.25 | *1/*1xN, *1/*2xN, *2/*2xN | metoprolol 代謝增加導致藥物濃度降低;然而,目前尚不清楚這是否會導致心率、血壓或臨床結果的臨床顯著變化。 | 無建議 由於缺乏關於 metoprolol 臨床效果降低的證據 | 無建議 |
| 正常代謝型(Normal metabolizer) | 1.25 ≤ x ≤ 2.25 | 2.25 2 1.75 1.5 1.25 | *2x2*10 *1/*1, *1/*2 *1/*10x3 *1/*17, *2/*29 *1/*10, *1/*41, *1/*9 | metoprolol 正常代謝 | 開始標準劑量 | 強烈建議 |
| 中間代謝型(Intermediate metabolizer) | 0 < x < 1.25 | 1 0.75 0.5 0.25 | *1/*5 *10/*17, *29/*41 *10/*10, *41/*41, *10/*41 *4/*10, *4/*41 | metoprolol 代謝減少導致藥物濃度增加;然而,這似乎不會轉化為心率、血壓或臨床結果的臨床顯著變化 | 開始標準劑量 | 中等建議 |
| 弱代謝型(Poor metabolizer) | 0 | 0 | *3/*4, *4/*4, *5/*5, *5/*6 | metoprolol 代謝減少導致藥物濃度顯著增加;這導致心率和血壓顯著降低,但對臨床結果的影響尚不清楚 | 以最低推薦起始劑量開始治療。仔細調整劑量至臨床效果或指南推薦劑量;更密切地監測心動過緩。或者,考慮選擇其他 beta-blocker。 | 中等建議 |
| 無法判定 | n/a | 攜帶一個或兩個 功能尚未確定 等位基因s 的個體 | *1/*22, *1/*25, *22/*25 | n/a | 無建議 | 無建議 |
a 請參閱 CYP2D6 基因資訊表 頁面上的 CYP2D6 等位基因頻率 表格以獲取 祖源-特定 等位基因 和 表現型 頻率。
b 等位基因 功能和 等位基因 活性值的分配,包括 等位基因 功能的引用,可以在 CYP2D6 基因資訊表 頁面上的 CYP2D6 等位基因 定義表和 CYP2D6 等位基因 功能表中找到。欲獲得 CYP2D6 雙倍型 和預測 表現型s 的完整列表,請參閱 CYP2D6 基因資訊表 頁面上的 CYP2D6 Diplotype to 表現型 表。
c 其中 xN 代表 CYP2D6 基因拷貝數。
d Metoprolol 通過 CYP2D6 沒有已知的活性代謝物。
e 評分方案在 指南補充的建議強度部分中描述。
metoprolol 途徑, 藥物動力學
概括
肝臟中metoprolol的代謝風格化描繪。
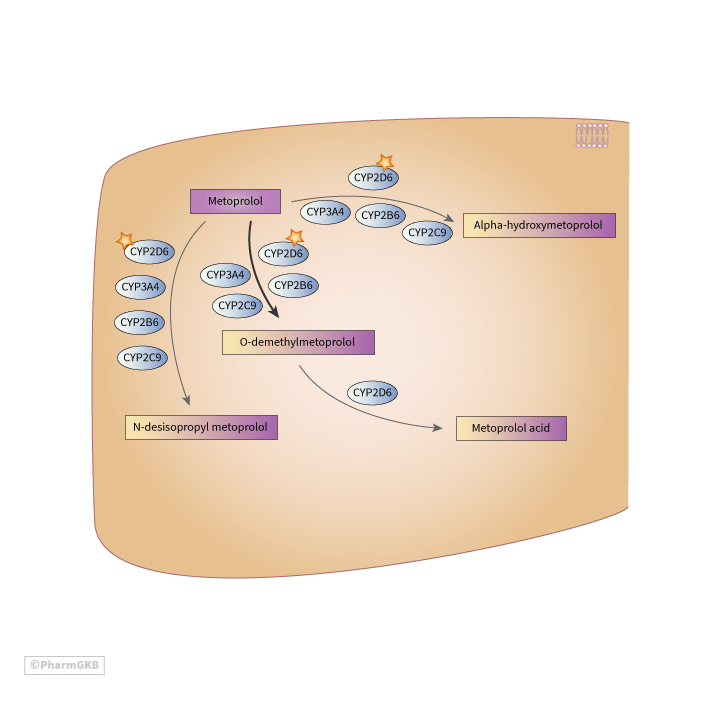
描述
metoprolol 是一種β-adrenergic blocker,適用於心衰竭和高血壓的治療。
metoprolol 經過廣泛的第一階段肝臟代謝 [Article:24648255]。metoprolol 及其代謝物的主要排泄途徑是通過腎臟,約有5%的劑量以母藥形式排泄於尿液中,其餘則以代謝物形式排泄。估計約100%的 metoprolol 代謝是通過 CYP 酶介導的,其中 CYP2D6 是關鍵的藥物基因,雖然不是唯一的基因 [Article:30087611][Article:16192109]。metoprolol 的代謝主要有三條途徑:O-去甲基化(約65%)、α-羥基化(10%)和N-去烷基化(10%)[Article:2222517][Article:30087611]。使用奎尼丁(quinidine),一種特定的 CYP2D6 抑制劑,顯示α-羥基化的殘餘通量仍為4%,N-去烷基化為8%,而O-去甲基化為19% metoprolol [Article:30087611]。
metoprolol 的主要代謝 途徑 是O-去甲基化 [Article:30087611][Article:25291152][Article:2222517]。這種代謝物性質上是瞬時的,並迅速氧化為 metoprolol acid,這是一種無活性的羧酸代謝物,也是人類尿液中主要的代謝物 [Article:25291152]。這條途徑對於R-metoprolol 旋光異構體是立體特異性的 [Article:2222517]。負責這一步驟的主要酶是 CYP2D6,然而在使用特定抑制劑的人的肝臟微粒體實驗中顯示,CYP3A4、CYP2B6 和 CYP2C9 在所有三條代謝途徑中均有輕微的貢獻 [Article:30087611]
α-羥基化產生了活性代謝物 alpha-hydroxymetoprolol,其體外效能約為 metoprolol 的十分之一 [Article:25291152][Article:30087611]。這個 途徑 對於-metoprolol 旋光異構體是立體特異性的 [Article:25291152]。
第三條途徑是N-去烷基化形成 N-desisopropyl metoprolol [Article:30087611]。
Beta-agonist/Beta-blocker 途徑, 藥效學
概括
氣道細胞中藥物作用於 β2 腎上腺素能受體之簡化藥效學途徑示意圖
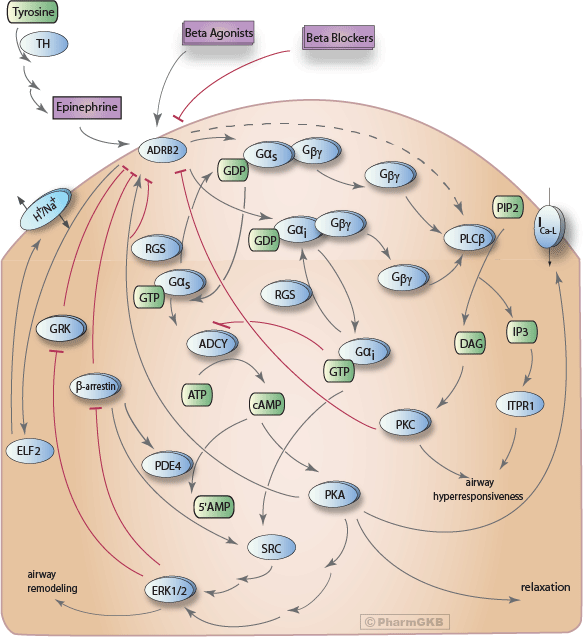
描述
當激動劑結合到β2腺苷酸受體(b2AR)時,會引發多種信號。b2AR的經典信號事件是其與異源三聚體G蛋白Gs的耦合。在非活性狀態下,Gs的α亞基與βγ亞基結合,並且GDP。當b2AR被激活時,Gαs從複合體中解離,GDP與GTP進行交換,並且Gαs激活腺苷酸酰化酶。該酶催化ATP轉化為cAMP。cAMP激活蛋白激酶A(PKA),該激酶磷酸化平滑肌細胞中的多種蛋白質,導致放鬆,或在氣道上皮細胞中增加纖毛的拍動。PKA也會磷酸化b2AR本身,從而降低其與Gs的耦合,這是一種去敏感化的形式。PKA-磷酸化形式的b2AR還促進與抑制性G蛋白Gi的耦合。解離的Gαi會抑制腺苷酸酰化酶。G蛋白異源二聚體的重組以及信號的停止,受到RGS蛋白的作用促進,這些蛋白加速GTP的水解。b2AR的功能部分受到幾種去敏感化機制的抑制,例如上述涉及PKA的負反饋迴路。該受體還會被幾個G蛋白偶聯受體激酶家族(GRKs)的成員磷酸化,這些成員不需要生成cAMP。GRK-磷酸化的b2AR作為β-逮捕素(b-arrestins)結合的底物,這些蛋白質在受體與G蛋白之間物理性地干擾並去敏感化功能。b2AR招募的β-逮捕素還作為其他蛋白質的運輸支架並啟動其他信號。β-逮捕素將磷酸二酯酶PDE4招募到受體/腺苷酸酰化酶微域。PDE4代謝cAMP,從而進一步去敏感化下游PKA-介導的事件。b2AR招募的β-逮捕素還啟動C-SRC的活化,最終導致ERK1和ERK2激酶的活化,這些激酶參與氣道重塑。Gαi可能也需要這一途徑,因為ERK1/2的活化部分被百日咳毒素阻斷。在某些細胞中,還存在一種嚴格PKA-依賴的途徑,導致ERK1和ERK2的活化。活化的ERK1和ERK2同時磷酸化GRK2和β-逮捕素,降低其功能,從而調節去敏感化。從G蛋白異源三聚體釋放的βγ也可以激活信號事件。βγ在激動劑促進的去敏感化過程中,招募肺部表達的主要GRK之一GRK2到b2AR。βγ還可以直接激活磷脂酶C的β亞型,導致肌醇-3磷酸(IP3)和二酸甘油酯(DAG)的生成。前者作用於IP3受體,釋放細胞內Ca++並增強收縮。DAG激活蛋白激酶C,該激酶磷酸化多種細胞蛋白,包括b2AR本身。這種βγ-PLC 途徑在氣道平滑肌的b2AR信號中被認為是次要的,但在Gibg增加的病理條件下可能會加強。在幾分鐘的激動劑暴露後,細胞表面的b2AR會經歷內化,這需要由β-逮捕素招募的AP2-夾克蛋白複合體。長期激活b2AR會通過未知機制增加b1亞型PLC的細胞表達,從而增加與M3-毒蕈鹼受體耦合的受體的收縮功能。激動劑對b2AR的激活還導致受體的泛素化,這一過程最終促進受體的降解。b2AR的泛素化是通過一種目前尚未識別的E3-泛素連接酶進行的,該酶由β-逮捕素招募到受體,而β-逮捕素本身則被E3-泛素連接酶Mdm2泛素化。激動劑結合到b2AR還通過直接與受體的細胞質尾部中的PDZ結構域相互作用,激活鈉氫交換調節因子(NERF)。(不需要G蛋白的激活或β-逮捕素的招募。)NERF調節參與細胞內離子調節的鈉氫泵。b2AR自發地在各種構象之間“切換”,包括那些有利於G蛋白耦合的構象,因此在任何時候,即使在沒有激動劑的情況下,細胞上的一些受體(相對於總數)也在進行信號傳遞。激動劑的結合有助於穩定受體在“活性”構象中,使得更多比例的受體參與信號傳遞。顯示了一些用於治療哮喘的常見激動劑,以及b2AR的內源性配體epinephrine。epinephrine合成的限速酶是酪氨酸羥化酶。還顯示了原型拮抗劑propranolol,以及用於治療心力衰竭的b1-b2AR拮抗劑carvedilol,這兩者均可結合到受體並阻止其與激動劑的接觸。